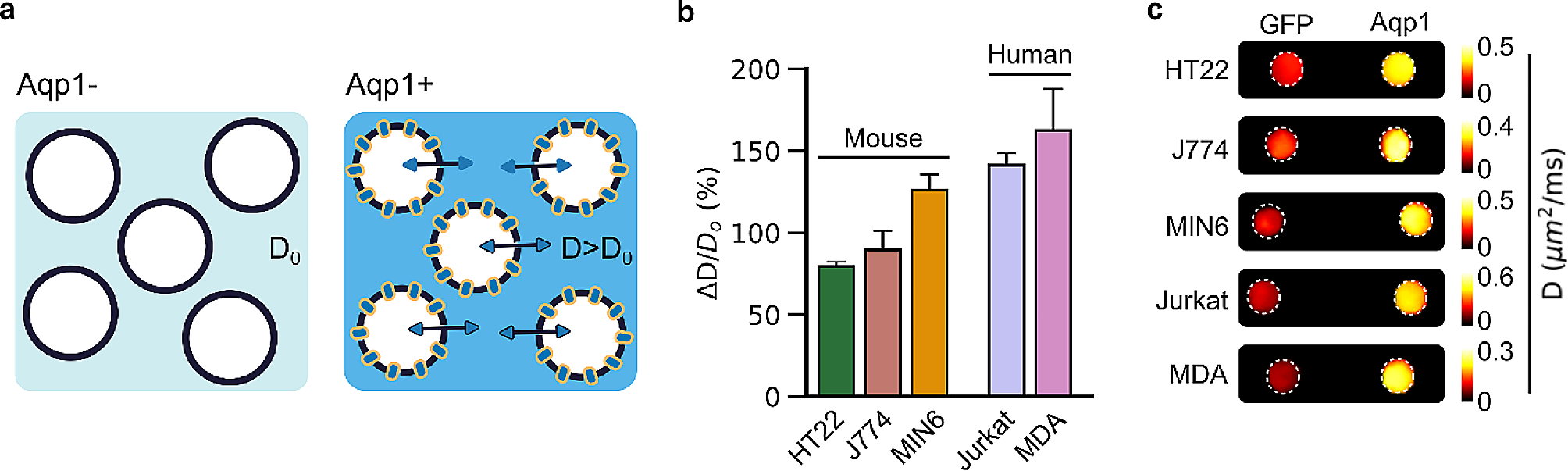
hiPSCs culture and RPE cell differentiation
The normal hiPSC cell line was cultured on Matrigel-coated plates(Corning, USA) using hiPSC medium (NuwacellTM hiPSC/hESC medium ncTarget; Nuwacell, Anhui, China). The hiPSCs were dissociated with 0.5 mM EDTA(Nuwacell) at 80% of confluence and passaged every 3–4 days. The culture medium was changed daily.
The hiPSC-RPE cells were differentiated as previously reported [27]. Briefly, hiPSCs were cultured in a differentiation medium composed of Dulbecco’s modified Eagle’s medium (DMEM, high glucose, Gibco,Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 50 μM β-mercaptoethanol (Sigma-Aldrich Corp., St. Louis, MO, USA), 1 × minimum essential medium–nonessential amino acids (MEM NEAA; Gibco), 1% penicillin-streptomycin (Gibco), and 20% of knockout serum replacement (KSR; Gibco) from D0-D42. 10 mM Nicotinamide (Sigma) was added to the differentiation medium at D0-D7, and 100 ng/ml Activin A (Peprotech, Thermo Fisher Scientific) and 3 µM CHIR99021 (Med Chem Express, New Jersey, USA) were added at D7-D14, D14-D42 respectively. On day 42, the pigmented cells were dissociated by TrypLE Express Reagent (Thermo Fisher Scientific) and enriched. Cells were then seeded on Matrigel-coated culture plates at a density of 3.5 × 104/cm2 in a maintained medium composed of high glucose DMEM, 4% KSR, 1 × MEM NEAA, 1% penicillin-streptomycin and 50 μM β-mercaptoethanol. The culture medium was changed every 2–3 days.
Generation and seeding of RPE spheroids
RPE cells were dissociated as described above. The collected hiPSC-RPE cells were seeded onto specific agarose micromolds as described previously [23] and incubated for 48 h. During this time, the suspended cells formed into a spheroid-like aggregate. After incubation, the RPE spheroids were seeded or bioprinted for further use.
Cell counting kit-8 assay (CCK-8)
A cell counting kit-8 (Bimake, Houston, USA) assay was performed to evaluate the viability of hiPSC-RPE cells. Briefly, RPE cells were seeded in a 96-well plate at a same density for 24 h. Then, Y27632 (10 μM) (Med Chem Express), Repsox (10 μM) (Med Chem Express) and a combination of Y27632 (10 μM) + Repsox (10 μM) were added into the culture medium, and the cells were further for additional 24 h. Then, 10 μL CCK-8 reagent was directly added into each well and the plate was incubated for 2 h until the color of medium turned orange. Subsequently, the absorbance value of the medium was read at 450 nm with a microplate reader.
Live/dead cell assay
The viability of RPE spheroids was evaluated using a live/dead cell imaging kit (Invitrogen, Thermo Fisher Scientific) following the manufacturer’s instructions. RPE spheroids were exposed to a 1 × Live/dead working solution for 25 min at 37℃ in the dark. RPE spheroids were imaged using a fluorescence microscope (Olympus, Japan). The percentage of live cells was acquired from three independent samples.
Relative quantification of pigmented area
To compare the expansion ability of RPE spheroids, the relative quantification process of the pigmented area was conducted with imageJ. The original color images of three groups were first combined into one image. Then, the combined images were converted to 8-bit grayscale images followed by the find edge process, threshold adjustment, target area setting, and the measurement of mean gray value (Fig. S6).
Flow cytometry
For the cell apoptosis experiment, hiPSC-RPE cells were dissociated, and the RPE suspension was divided evenly and treated with Y27632 (10 μM), Repsox (10 μM) and Y27632 (10 μM) + Repsox (10 μM), respectively. The Annexin V-Alexa Fluor 488/PI Kit (4A biotech, Beijing, China) was used. First, the cell suspension was centrifuged, washed with PBS, then centrifuged again. Second, the RPE cells were re-suspended in 1 × binding buffer and incubated with annexin V-Alexa Fluor 488 for 5 min. Third, propidium iodide solution was added to the cell samples, and the samples were detected immediately and analyzed using a flow cytometer (Novocyte).
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 15 min at room temperature and rinsed thrice with PBS. Following that, the cells were permeabilized in 0.1% Triton X-100 in PBS for 10 min and blocked with 3% BSA in PBS for 1 h at room temperature. Next, the cells were incubated with primary antibodies overnight at 4℃. After rinsing with PBS, the cells were stained with Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific, 1:1000) for 1 h at room temperature. Following PBS wash, the cells were stained with DAPI for 5 min and mounted with a mounting medium. Immunofluorescence was examined using a fluorescence microscope (Olympus, Japan). CoraLite594-Phalloidin (Proteintech, USA) conjugates were used to label filamentous actin (F-actin). The other antibodies used are listed in Table S1.
Reverse Transcription and Quantitative Real-time PCR (RT-qPCR)
Total RNA was extracted using the FastPure® Cell Total RNA Isolation Kit (Vazyme, Nanjing, China) and dissolved in RNase-free water. RNA samples were quantified by measuring the OD value at 260 nm, and the OD 260/280 ratios for all RNA samples fell within the range of 1.8 to 2.1. Reverse transcription was performed using the HiScript III RT SuperMix for qPCR (+gDNA wiper) kit (Vazyme), following the manufacturer’s instructions. RT-qPCR was conducted using the ChamQ Universal SYBR qPCR Master Mix kit (Vazyme), and the PCR mixture was run in the CFX96 Real-Time PCR system (Bio-Rad, USA). Three technical replicates were set and expression levels were normalized to the expression of GAPDH. Relative expression levels compared to the control group were determined by calculating the 2 - ΔΔCt.
Western blotting
The cells were washed twice with PBS, then treated with RIPA buffer (Thermo Fisher Scientific), and centrifuged at 12,000 rpm for 15 min. The supernatant was reserved. The protein concentration of the samples was determined using a BCA protein assay kit (Thermo Fisher Scientific). The protein samples were adjusted to a concentration of 2 μg/μl. Subsequently, 10 μl of the protein samples were loaded and separated by electrophoresis on a 10% SDS-PAGE gel (SDS-PAGE gel kit, Solarbio Life Sciences, Beijing, China). Following protein separation, the proteins were transferred to PVDF membranes (Millipore, Billerica, MA, USA), blocked with 5% BSA at room temperature for 1 h, and then incubated with primary antibodies overnight at 4℃. The membrane was washed three times with TBST and subsequently incubated with HRP-conjugated secondary antibodies for 2 h at room temperature. After further washing with TBST, protein bands were visualized using an ECL kit (Millipore, USA) with a gel imager. The antibodies used are listed in Additional file 3, Table S1.
Karyotype analysis
The karyotype analysis were conducted using a standard G-band technique (350G–400G) and analyzed by Ikaros karyotyping system. 20 metaphases were examined. The number of chromosomes as well as the presence of structural chromosomal abnormalities was examined.
Transepithelial electrical resistance (TER) assay
TER assay was conducted using the EVOM2 voltohmmeter (World Precision Instruments, Sarasota, Florida, USA), following the manufacturer’s instructions. RPE cells and RPE spheroids were seeded onto PET Transwell membranes (3 μm pore size, Corning) coated with Matrigel at a same density and cultured for 2 weeks. TER values were measured from three replicates on day 14. Background resistance was determined from a blank culture insert. The TER value (Ωcm2) was calculated using the following formula.
$$\mathrm(\Omega /}}^) = (}}_}}-}}_}})/}$$
Rtotal represents the total resistance measured (Ω), Rinsert represents the resistance of the blank insert, and A represents the membrane area (cm2) of the insert.
Phagocytosis assay
POS was obtained from fresh porcine eyes, isolated in sucrose gradient solution, and centrifugated at 106,000 × g for 50 min at 4 ℃, following established protocols [59, 60]. Then, POS was labeled with FITC isomer 1 (0.5 mg/ml) in DMEM for 1 h at room temperature in the dark. RPE cells were exposed to the labeled POS solution containing 10%FBS for 6 h at 37℃. After incubation, RPE cells were thoroughly washed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 3% BSA. Subsequently, the ZO-1 antibody was used for immunolabelling, and nuclei were stained with DAPI. Imaging was performed using a confocal microscope (Leica TCS SP8, Germany).
RNA-sequencing analysis
Total RNA was isolated from hiPSC-RPE in 2D culture and spheroid culture way with Trizol total RNA reagent (Invitrogen). RNA sequencing was performed by the DNBSEQ platform (BGI Tech, Shenzhen,China). After the filtration of raw data, the clean reads were generated and aligned to the human genome assembly. After that, the qualified reads were standardized and quantified with transcripts per kilobase million(TPM) values. Differentially expressed genes(DEGs) were identified between the two groups. The significance of DEGs was defined by the combination of |log2FoldChange|≥ 1 and Q-value ≤ 0.05. Gene Ontology (GO) terms enrichment and the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were performed to further analyze data.
Fabrication of collagen vitrigel (CV) membrane
The CV membrane was constructed as previously described [61]. Briefly, 5 mg/ml pre-cooling type I collagen solution (extracted from bovine tendon, Guangzhou Trauer Biotechnology, China) and 10 × DMEM (Hyclone, USA) were mixed in volume of 1:9. The mixed collagen hydrogel was centrifugated (2000 rpm, 3 min) and added into 6-well plates. The plates were then incubated at 37℃ to complete the gelation of the collagen for 30 min and dried in a clean air chamber at 10℃ and 40% humidity for 8 h to roughly remove the moisture of the gel. After that, the collagen gel was dried on a clean table and exposed to ultraviolet rays at room temperature for additional days to achieve vitrification. Then, the vitrified membrane was rehydrated with PBS. Using tweezers, the CV membrane was gently removed and placed into a ring-shaped polytetrafluoroethylene mold for the subsequent loading of RPE cells.
Scanning electron microscope (SEM)
SEM was used to examine the ultrastructural surface of the CV membrane and the RPE cells cultured on the CV membranes. RPE spheroids were initially seeded onto the CV membrane and cultured for 14 days. Subsequently, the cell sample and CV membrane were fixed in 2.5% glutaraldehyde for 2 h, followed by three washes with a PBS solution. The cell samples were then dehydrated using a series of increasing ethanol concentrations in PBS (50%, 70%, 80%, 90%, 95%, 100%, 100%). Afterward, the samples underwent critical point drying and gold metal coating, and were imaged using a scanning electron microscope (Zeiss, Ultra 55).
Automatic RPE spheroids bioprinting
The automatic spheroids bioprinting device was designed and assembled by our team. Its precision and accuracy were validated through calibration and testing conducted by a certified center (Ceprei, China; Certificate NO. 1GA18004742-0002). RPE spheroids from 96-well U-shaped plates were aspirated and accurately printed onto the surface of CV membranes automatically following a preset pattern (Movie S1). Subsequently, culture medium was gently added to the cell plates, and they were placed in an incubator at 37℃ with 5% CO2 for further cultivation.
Statistical analysis
The data are presented as means ± standard deviation (SD) and derived from a minimum of three samples. GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA) and ImageJ (National Institutes of Health, Bethesda, MD, USA) were employed for data analysis. The unpaired two-tailed t-test was applied for comparison between groups. P values < 0.05 were considered statistically significant.



留言 (0)