This clinical trial (NCT 03695250) was conducted following all applicable regulatory requirements and was approved by the UC Davis Institutional Review Board (IRB).
Patient selection
Eligible patients were age ≥ 18 years with unresectable or metastatic, histologically or imaging-confirmed hepatocellular carcinoma (HCC) which was not amenable to curative treatment approach. Patients must have had measurable disease by RECIST v.1.1 criteria, and ≥ 1 liver lesions accessible for core biopsy that was either untreated with prior liver-directed therapy or progressed following liver-directed therapy. Patients had to have Child-Pugh score A at enrollment, Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, and able to swallow intact pills. Patients with active hepatitis B virus (HBV) were permitted if antiviral therapy for hepatitis had been administered for ≥ 8 weeks and viral load was < 100 IU/mL prior to the first dose of trial treatment; patients with untreated hepatitis C virus (HCV) were permitted. Other eligibility criteria included: absolute neutrophil count (ANC) of ≥ 1000 cell/mm3, platelet count of ≥ 50,000/mm3, hemoglobin ≥ 8 g/dL, total bilirubin and creatinine ≤ 2x the institutional upper limit of normal (ULN), and aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase < 5x the institutional ULN. Patients had to undergo mandatory pre-treatment biopsy in cases where insufficient archival tumor specimen was available as well as on-treatment biopsy.
Key exclusion criteria included receipt of any current or prior systemic cancer-related therapy, immunodeficiency history, active autoimmune disease or diseases/disorders requiring a systemic steroid equivalent of prednisone ≥ 10 mg/day or immunosuppressive therapies given within 7 days before the first dose of the study, any active bacterial, fungal or viral infections (excluding HBV and HCV), and any known history of pneumonitis. Patients could not receive liver directed therapy within 4 weeks of the first dose of study drug. Patients were also ineligible if there was clinically significant ascites, hepatic encephalopathy, esophageal/gastric varices with bleeding within 3 months of study enrollment, receipt of live attenuated vaccines within 30 days of first study treatment, additional malignancies, G6PD deficiency or other congenital/autoimmune hemolytic disorders, history or presence of cytochrome b5 reductase deficiency, QTc interval > 480ms and any treatment with botanical preparations within two weeks prior to randomization.
All subjects provided written consent and the study was compliant with Good Clinical Practices guidelines and the Declaration of Helsinki.
Study design and objectives
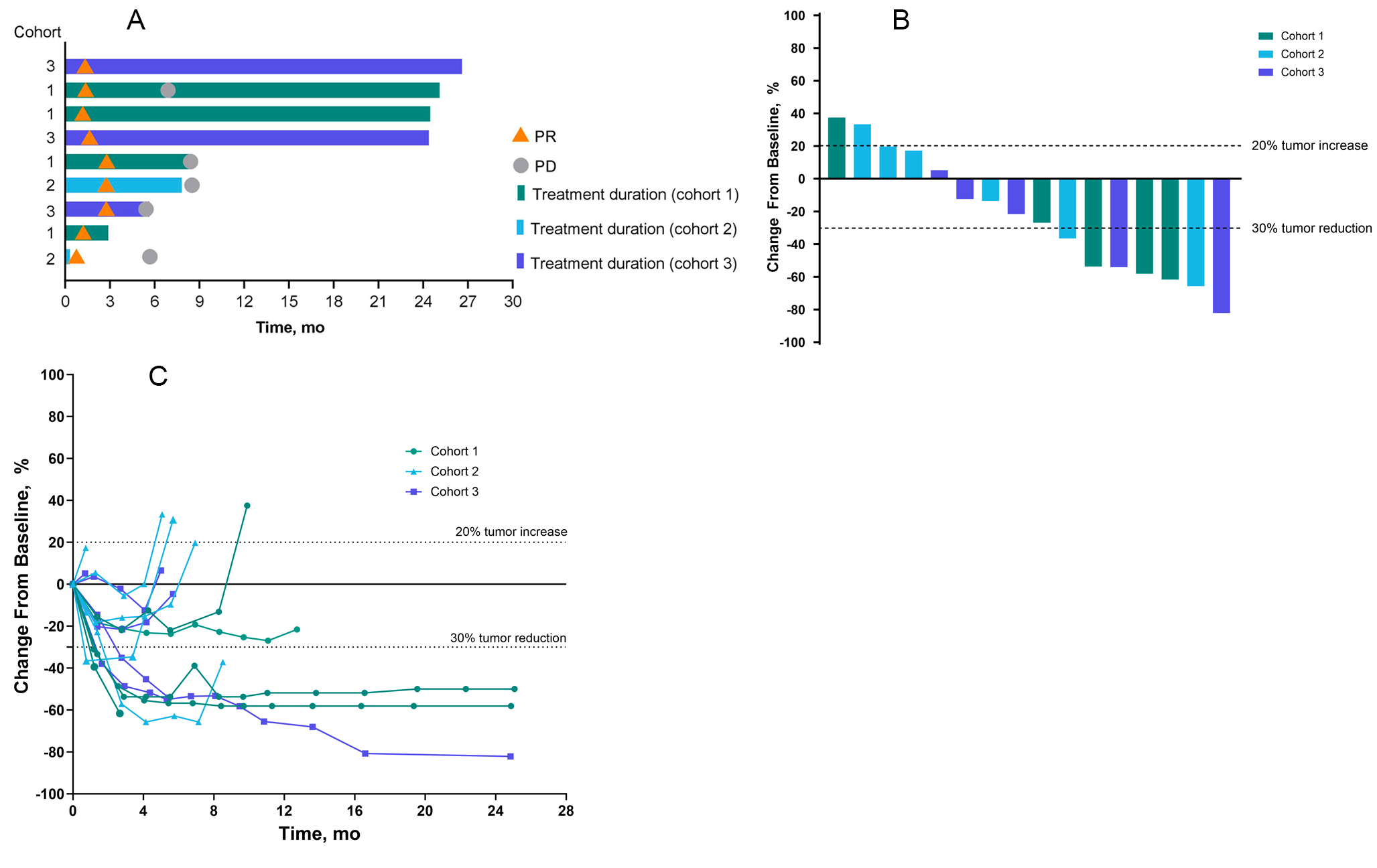
This was an open-label Phase I/II clinical trial. Phase I was conducted as a 3 + 3 dose escalation study and Phase II was planned as a Simon two-stage design to allow for early trial stoppage for futility. The primary objectives of this study were to determine the safety and tolerability of BMS-986,205 in combination with nivolumab in unresectable/metastatic HCC in the first line setting using CTCAE V5.0 criteria, and to determine efficacy of this combination using RECIST (version 1.1) criteria. Secondary objectives included obtaining preliminary data on disease control rate (DCR), duration of response (DOR), ORR using immune RECIST (iRECIST) criteria, progression free survival (PFS), and OS, and to further evaluate the safety of this combination.
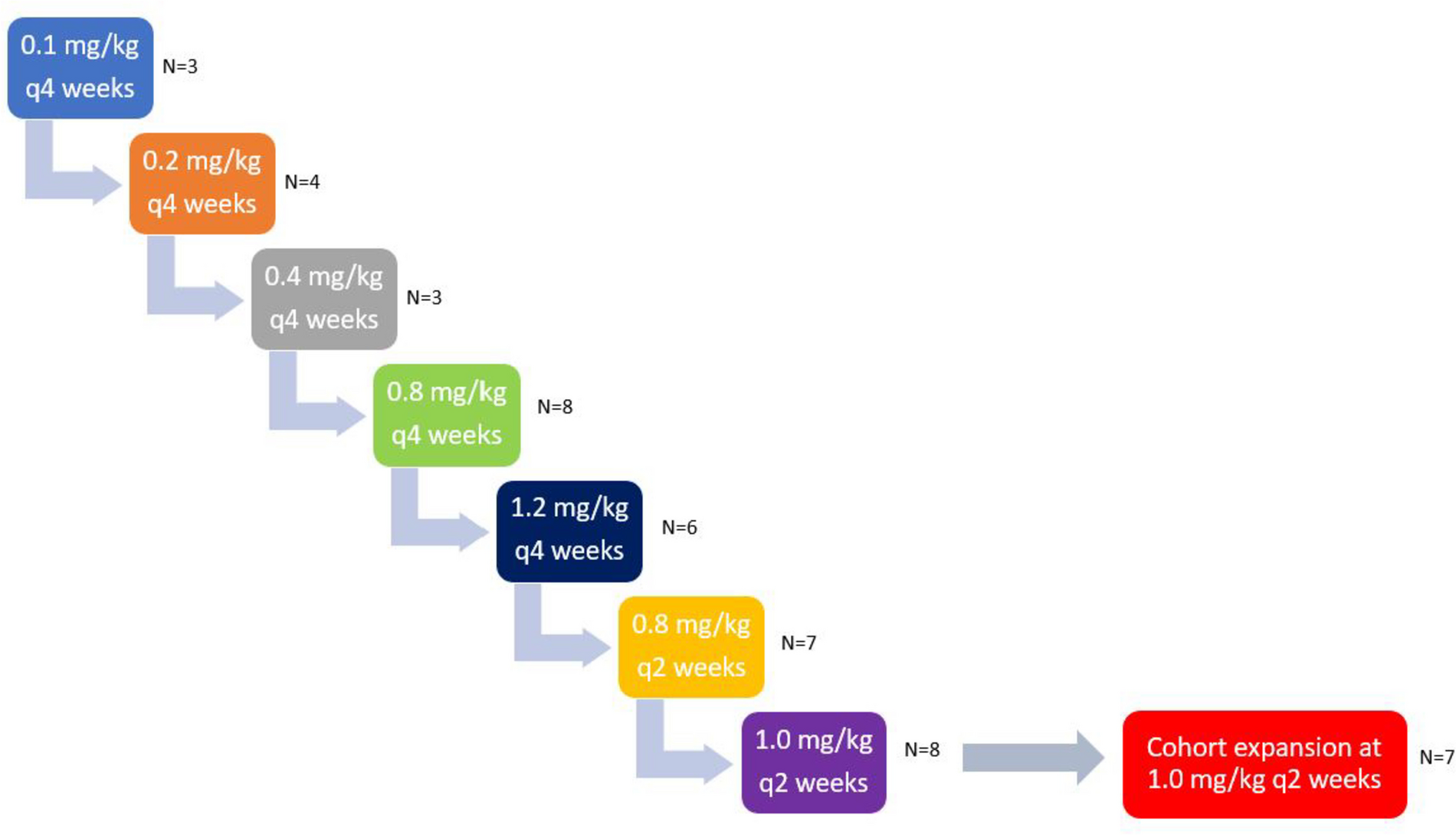
For the Phase I dose-escalation portion, the primary endpoint was to determine the safety profile of BMS-986,205 in two dose levels (50 mg and 100 mg) with fixed-dose nivolumab 240mg/m2 IV every 14 days. Patients were treated following a standard 3 + 3 design, with dose level 1 (DL1) defined as BMS-986,205 50 mg daily and dose level 2 (DL2) as BMS-986,205 100 mg daily, in order to determine the maximum tolerated dose (MTD). MTD was defined as the dose at which ≤ 1 out of 6 patients develops a DLT at BMS-986,205 DL1 or DL2. DLT was defined as any drug-related Grade 2 uveitis or eye pain requiring systemic therapy or unresponsive to topical therapy and failing to improve within 2 weeks of starting therapy, any Grade 2 drug-related pneumonitis or interstitial lung disease unresponsive to dose delay and systemic steroids within 2 weeks, any Grade 3 non-skin drug-related adverse event (excluding laboratory abnormalities, fatigue and nausea) unrelieved or controlled with appropriate care within two weeks, any Grade 4 drug-related adverse events including laboratory abnormalities (except Grade 4 leukopenia or neutropenia) lasting < 14 days, methemoglobin levels ≥ 15% and Grade ≥ 3 hemolysis (i.e., requiring transfusion or medical intervention such as steroids), and any study drug-related Grade ≥ 3 hemolysis requiring transfusion or steroids. The following drug-related hepatic function laboratory abnormalities were also considered DLTs: AST or ALT more than 10 times the upper limit of normal for more than two weeks, AST or ALT more than 15 times the upper limit of normal (irrespective of duration), and total bilirubin greater than five times the upper limit of normal (irrespective of duration). To be evaluable for DLT, the patient must have received at least 75% of the total intended dose of BMS-986,205 during the first 6 weeks of therapy. Therapy was continued until progression or 24 months, with the option to continue therapy after 24 months per treating physician discretion.
The Phase II portion was planned to be a Simon two-stage design with a total of 17 patients, inclusive of 6 patients from the Phase I portion treated at the MTD.
Safety and efficacy assessments
Safety assessments consisted of monitoring and recording all adverse events, including serious adverse events, lab parameters and physical exam at each study visit. Toxicity was evaluated according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE, version 5.0 criteria). All patients receiving any amount of study drug were evaluable for toxicity.
Response was assessed using computed tomography (CT) scans of chest, abdomen, and pelvis at baseline and then at the end of 8 weeks, 16 weeks, and then every 12 weeks per Response Evaluation Criteria in Solid Tumors (RECIST) guideline (version 1.1) as well as immune RECIST (iRECIST) criteria. DOR, PFS and OS were evaluated using Kaplan-Meier plots.
Adverse events were summarized according to organ system, laboratory category, and dose level in frequency tables graded according to CTCAE v5.0. Information regarding each subject’s course including completion of therapy, dose delays, premature discontinuation, and major protocol violations were tabulated and summarized.
Statistical considerations
This was a phase I/II study with a planned Simon optimal two-stage design for efficacy evaluation in Phase II. In Phase I, patients were treated following a standard 3 + 3 design with dose escalation in BMS-986,205 and fixed-dose nivolumab. In Phase II dose expansion, initially three additional patients were to be enrolled at the MTD for a total of 9 patients at the MTD, including the 6 patients treated at the MTD in the Phase I portion. An interim efficacy analysis was to be done of those 9 patients and if ≥ 1 of 9 initial patients treated at MTD achieved a response, 8 more patients would be accrued at the MTD. If ≥ 3 of 17 patients treated at MTD achieved a response, the treatment would be deemed worthy of further study, provided the safety profile is acceptable. The Simon optimal two-stage design provided 80% power to detect the difference between an acceptable response rate by RECIST v1.1 of 25% vs. an unacceptable rate of 5% at the 0.05 level (1sided). ORR was estimated as the proportion of participants who experienced an objective response, along with its exact 95% confidence interval. DCR was analyzed similarly. DOR, PFS, and OS were analyzed using Kaplan-Meier methods, and medians and 95% confidence intervals were computed. Safety analyses were tabulated for each patient and summarized in frequency tables.

留言 (0)