This study was done in accordance with local and/or national regulations and the ethical principles in the Declaration of Helsinki. An institutional review board or independent ethics committee approved the study protocol before the study was initiated at each site. Patients provided written informed consent before entering the study.
Patients
Eligible patients were ≥ 18 years of age and had histologically or cytologically confirmed advanced or metastatic solid tumors with progression on, or intolerance to, all other treatments known to confer benefit. Other inclusion criteria included measurable disease as determined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, an Eastern Cooperative Oncology Group performance status of 0 or 1, and adequate organ function. Patients were ineligible if they had received chemotherapy, definitive radiation, or biological cancer therapy ≤ 4 weeks (≤ 2 weeks for palliative radiation) prior to the first dose of study treatment or if they were still experiencing grade ≥ 2 adverse events from a cancer therapy administered > 4 weeks earlier. Other exclusion criteria included clinically active central nervous system metastases, carcinomatous meningitis, active infection requiring therapy, history of noninfectious pneumonitis requiring steroids or current pneumonitis, active autoimmune disease requiring systemic treatment in the last 2 years, diagnosis of immunodeficiency or receipt of immunosuppressive therapy ≤ 7 days before treatment allocation, and grade > 1 peripheral neuropathy or paresthesia at baseline.
Study design
This was a phase 1b, open-label, multicenter, multiple-dose, dose-escalation study. Patients received pembrolizumab 200 mg intravenously every 3 weeks in combination with oral selumetinib. The recommended phase 2 dose for selumetinib monotherapy is 75 mg orally twice daily administered on a continuous basis [14]. At this dose, enhanced MEK inhibition is proposed to be associated with greater response, as well as an escalation in dose-dependent adverse events. To mitigate dose-dependent adverse events associated with MEK inhibition, this study chose an intermittent dosing schedule for selumetinib (2 weeks on/1 week off) and began treatment at a dose below the recommended phase 2 dose. The starting dose of selumetinib was 50 mg twice daily on days 1 to 14 of each 3-week treatment cycle, which was escalated in 25 mg increments up to a prespecified maximum of 300 mg twice daily on days 1 to 14 of each cycle. Treatment was continued until documented radiographic disease progression, as assessed by the investigator per RECIST version 1.1 and then confirmed by modified RECIST for immune-based therapeutics (iRECIST) [15], unacceptable toxicity, intercurrent illness, investigator decision, or completion of 35 treatment cycles (~2 years). Patients with unconfirmed disease progression may have continued treatment at the discretion of the investigator until progression was confirmed using iRECIST; if repeat imaging did not confirm progressive disease per iRECIST, as assessed by the investigator, and the patient continued to be clinically stable, study treatment could have continued.
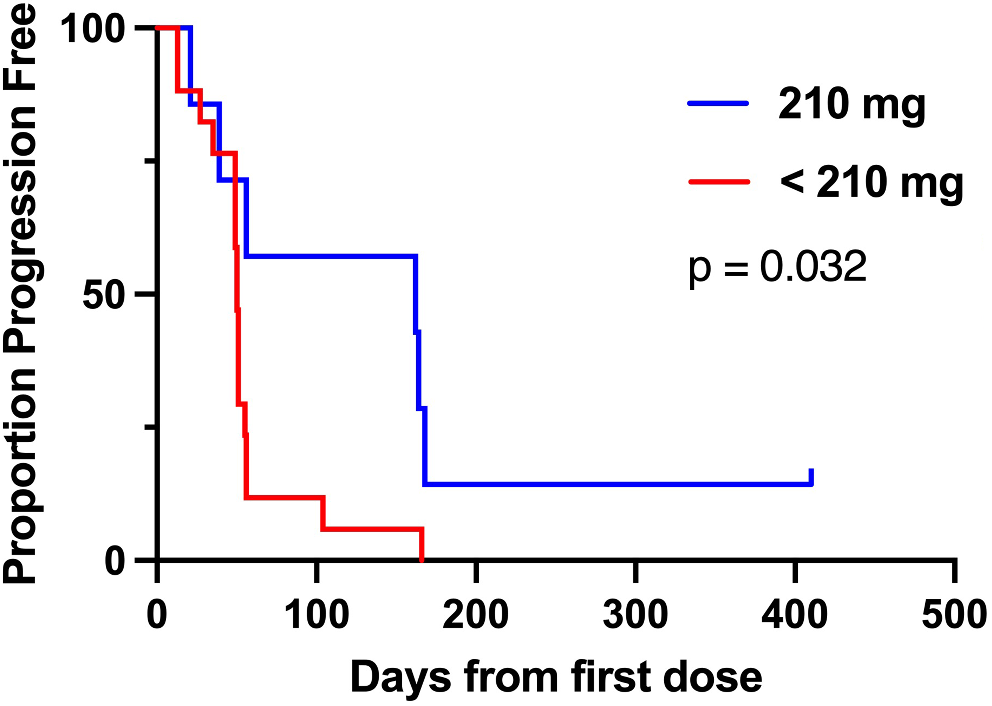
At least 3 patients were enrolled at each dose level; the modified toxicity probability interval (mTPI) design [16], with a target dose-limiting toxicity (DLT) rate of ~30%, was used to identify a potential maximum tolerated dose. Dose escalation and de-escalation decisions were based on the mTPI design and were dependent on the number of patients enrolled and number of DLTs observed at the particular dose level (Online Resource 1). Dose finding was considered complete after 14 patients were enrolled at any of the tested dose levels and the decision was made to stay at that dose level.
Assessments
Patients were monitored for DLTs (see Online Resource 2 for definition) during the first 21 days of treatment. Adverse events were monitored throughout the study and for 30 days after cessation of study treatment (90 days for serious adverse events) and were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. A full ophthalmic examination was performed on day 1 of cycle 2 and every 8 weeks thereafter through treatment discontinuation, as selumetinib monotherapy is known to cause ocular toxicity, including blurred vision, photophobia, cataracts, and ocular hypertension [3, 17, 18].
Plasma samples for analyzing the pharmacokinetics of selumetinib were collected on day 1 of cycle 1 (predose; 1, 2, 4, and 6 h postdose; and 8–12 h postdose [before evening dosing]). Additional predose samples were collected on days 1 and 14 of cycle 2 and on days 1 and 14 of cycle 5.
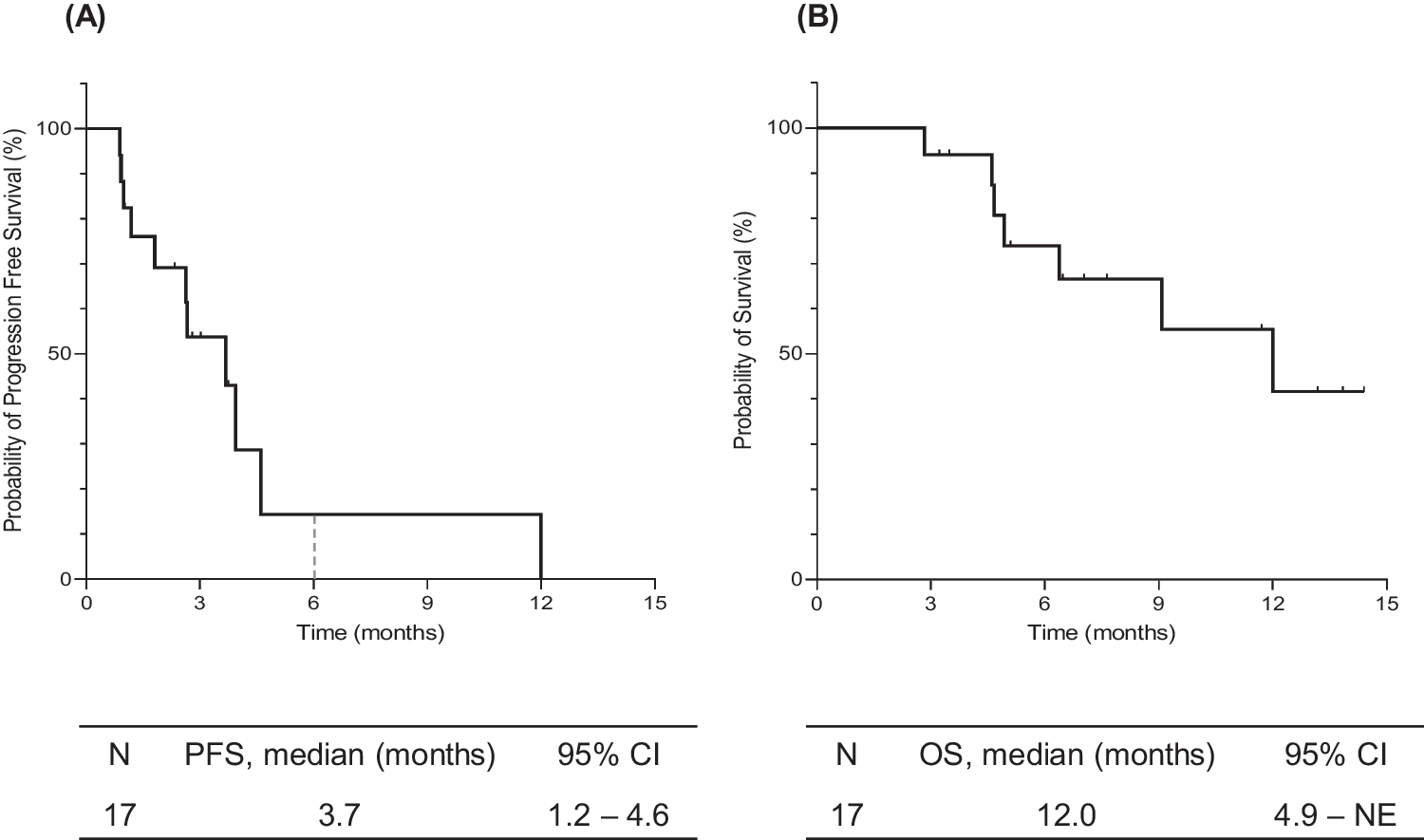
Tumor imaging was performed at screening, every 9 weeks for 12 months, every 12 weeks during months 12 to 24, and at treatment discontinuation. For patients who discontinued study treatment without disease progression, an effort was made to continue tumor imaging (same schedule as during treatment) until the start of new anticancer treatment, disease progression, pregnancy, death, withdrawal of consent, or the end of the study.
Biomarkers were assessed at baseline and included PD-L1 combined positive score (CPS), T-cell‒inflamed gene expression profile (Tcellinf GEP), tumor mutational burden (TMB), microsatellite instability (MSI), and KRAS/BRAF mutations [19,20,21].
Endpoints
Primary endpoints were the occurrence of DLTs, adverse events, and study treatment discontinuations due to adverse events. Pharmacokinetic parameters of selumetinib were assessed as secondary endpoints. An exploratory endpoint was the objective response rate (ORR) per RECIST version 1.1 as assessed by the investigator, defined as the proportion of patients with a confirmed complete or partial response. Biomarkers were an additional exploratory endpoint.
Statistical analysis
The sample size was planned at 50 to 84 patients and depended on the observed DLT rate. Analyses of adverse events were based on the safety population, which included all patients who received ≥ 1 dose of study treatment. The DLT population included all patients in the safety population who completed cycle 1 without a DLT or who experienced a DLT in cycle 1. Estimates of DLT rates across dose levels in each treatment arm were analyzed using isotonic regression with the pooled adjacent violators algorithm [16]. Pharmacokinetic analyses were based on all patients who complied with the protocol sufficiently to ensure that their data would likely reflect treatment effects. Efficacy analyses were based on all patients with a baseline scan who had measurable disease by investigator assessment and who received ≥ 1 dose of study treatment. No formal hypothesis testing was planned.

留言 (0)