
記住我

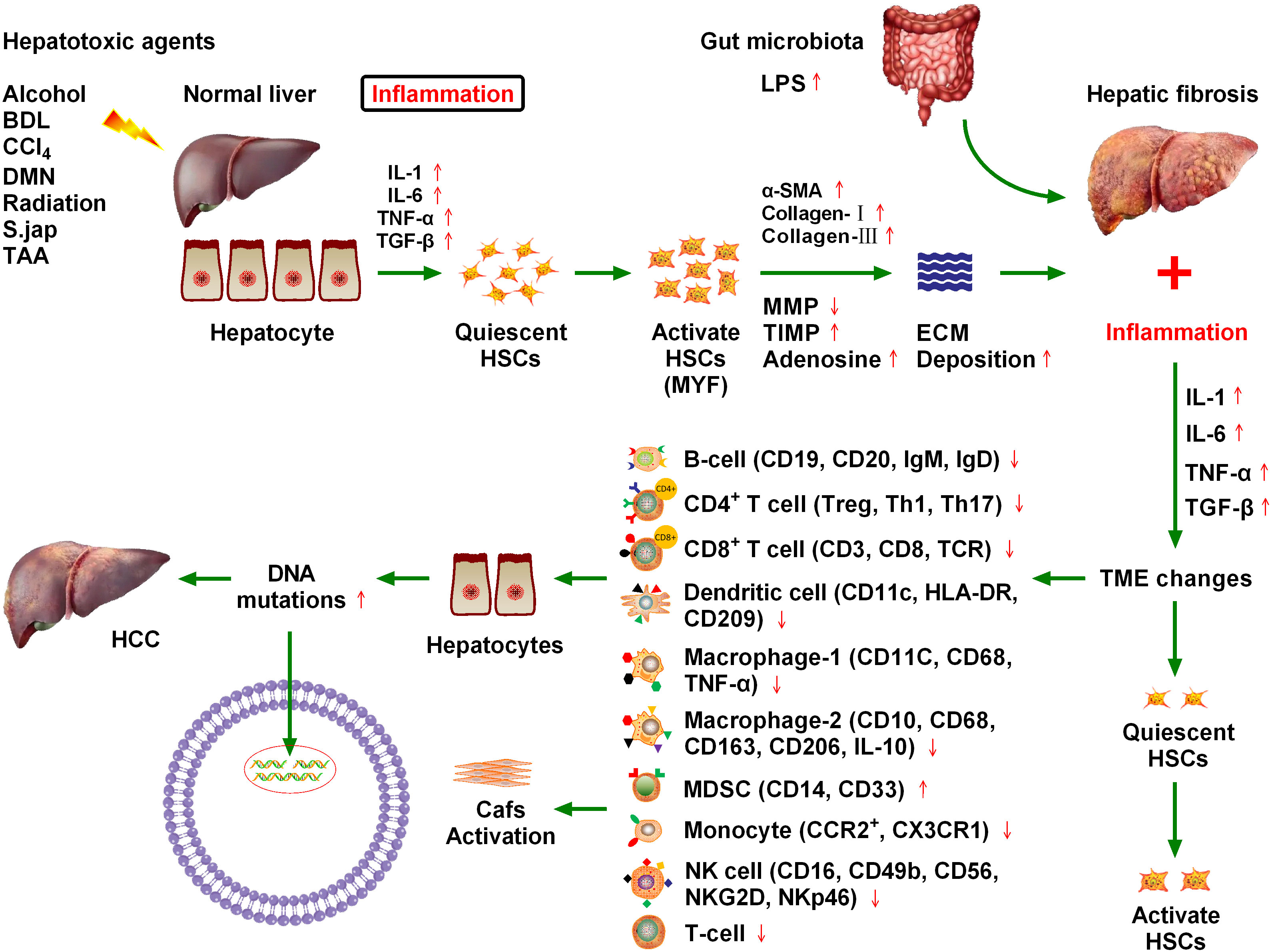
HCC is the fifth most common cancer worldwide and the third leading cause of cancer death (1). Recent epidemiological data indicate that death rates from HCC are increasing in the United States and Europe, and, together with incidence, are expected to double in the next 10–20 years (2). HCC can be primary or secondary. Primary HCC includes HCC and intrahepatic cholangiocarcinoma (ICC), mixed HCC, and bile duct cell carcinoma. HCC and ICC are the two main types of primary HCC (3). HCC is the most common primary liver cancer and the fourth leading cause of cancer-related death globally, accounting for more than 90% of all primary HCC cases. Chronic hepatitis B or C virus infection is the main cause of HCC, while other causes include alcoholism, autoimmune liver disease, and nonalcoholic steatohepatitis (4). Constant inflammation damages deoxyribonucleic acid (DNA) in regenerating liver cells, which leads to genetic changes that increase the chance of cancer development (5) (Figure 1). Chronic hepatitis infection or long-term liver injury often leaves the liver in a state of chronic inflammation (6, 7). Moderate inflammation can fight pathogens and repair tissue damage in liver, however, persistent liver inflammation can disrupt the microenvironment and tip the balance in favor of liver carcinogenesis (8, 9). Over the past decade, new researches have demonstrated that the immune microenvironment plays critical roles in HCC progression, and therapies targeting tumor microenvironment (TME) have been reported to effectively inhibit HCC growth in both animal models and clinical trials (6–9).
Figure 1 Chronic cytokine stimulation of liver cells leads to genetic mutations that induce hepatocellular carcinoma (HCC), and ultimately hepatic fibrosis. Various injury factors, including viral infection, alcohol, obesity, and insulin resistance, act on the liver, causing liver inflammation, an immune response, hypoxia, oxidative stress, hepatocyte necrosis, and apoptosis. Injury and inflammation cause regeneration of hepatocytes and activation of hepatic stellate cells (HSCs), which are the main sources of liver scarring. After activation, a series of phenotypic and biological behavior changes occur in HSCs, including the promotion of fibroplasia and migration and the release of many cytokines, which promote proliferation and angiogenesis, and prevent apoptosis. Inflammatory cells, such as macrophages, natural killer cell lymphoma and leukemia, T lymphocytes, and B lymphocytes, may be involved in liver injury and fibrosis. Chronic inflammation and regeneration processes lead to dysregulation of hepatocyte growth, genomic instability, DNA damage, dysplasia, and malignant transformation, which ultimately lead to HCC. The immune microenvironment composed of different immune cells plays a key role in HCC progression.
1.2 Hepatic fibrosisNumerous inflammatory factors continue to stimulate hepatic stellate cells, leading to their activation, which is the main cause of hepatic fibrosis (10). Approximately 80% of HCC cases occur in the context of chronic inflammation and cirrhosis caused by viral hepatitis. The hepatic inflammation and fibrosis environment play an important role in the development of HCC, and the treatment and prognosis of HCC are also complicated by the tumor stage and the degree of liver dysfunction (11). Various injury factors, including viral infection, alcohol, obesity, and insulin resistance, act on the liver, which can cause liver inflammation, immune response, hypoxia, oxidative stress, hepatocyte necrosis, and apoptosis (12). Injury and inflammation cause the regeneration of hepatocytes and the activation of HSCs, which are the main sources of liver scars. After activation, HSCs undergo a series of phenotypic and biological behavior changes, including fibroplasia promotion, migration, and release of many cytokines that promote proliferation, angiogenesis, and anti-apoptosis (13). Inflammatory cells such as macrophages, natural killer (NK) cells, T lymphocytes, and B lymphocytes may be involved in liver injury and fibrosis. Chronic inflammation and regeneration processes lead to the loss of hepatocyte growth, regulatory genomic instability, DNA damage, dysplasia, and malignant transformation, ultimately leading to the development of HCC (13) (Figure 1).
1.3 Liver inflammation and the fibrosis microenvironment promote HCC progressionA close relationship exists between chronic liver inflammation, hepatic fibrosis, and HCC. Most chronic liver diseases are characterized by diffuse chronic inflammation, necrosis, and fibrosis. Chronic inflammation and fibrosis is a dynamic process of accumulation of lymphocytes, macrophages, and matrix cells that undergo secretory and paracrine interactions (11). Inflammatory cells belonging to innate immunity (e.g., NK cells and macrophages) and adaptive immunity (e.g., T lymphocytes and B lymphocytes) are involved in hepatic injury and fibrosis. By contrast, injured hepatocytes, Kupffer cells, and HSCs are involved in inflammation induction. Matrix cells can regulate the differentiation and function of antigen-presenting cells. The pattern of cytokine and chemokine secretion in the matrix determines T lymphocyte migration and polarization (14). Tumors may occur when chronic inflammation and injury healing processes become dysregulated. Malignant transformed hepatocytes may replace proliferative nodules, which are atypically altered during regeneration. In addition, hypoxia and inflammation are major factors that stimulate the proliferation of blood vessels and promote tumor growth (Figure 1). Inflammatory signals such as toll-like receptor 4 (TLR4) and nuclear factor kappa B (NFκB) promote tumor cell proliferation and migration by producing numerous cytokines and altering matrix, chemotactic growth, and lymphovascular hyperplasia factors (15). Because of the close relationship between hepatic fibrosis and HCC, it is crucial to explore the influence of fibrosis signaling on the occurrence, development, recurrence, and metastasis of HCC to improve the standard of HCC prevention and treatment (16).
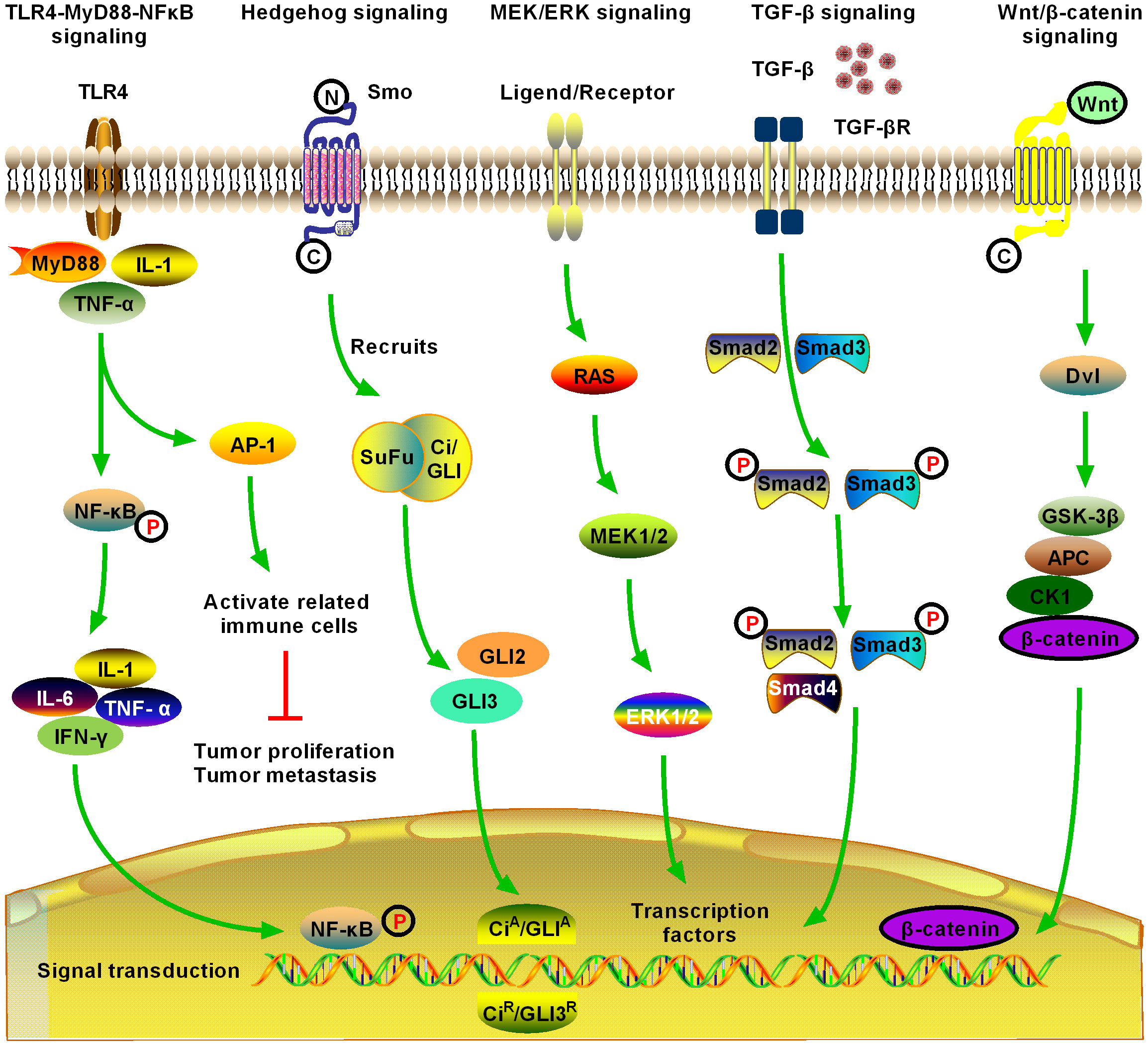
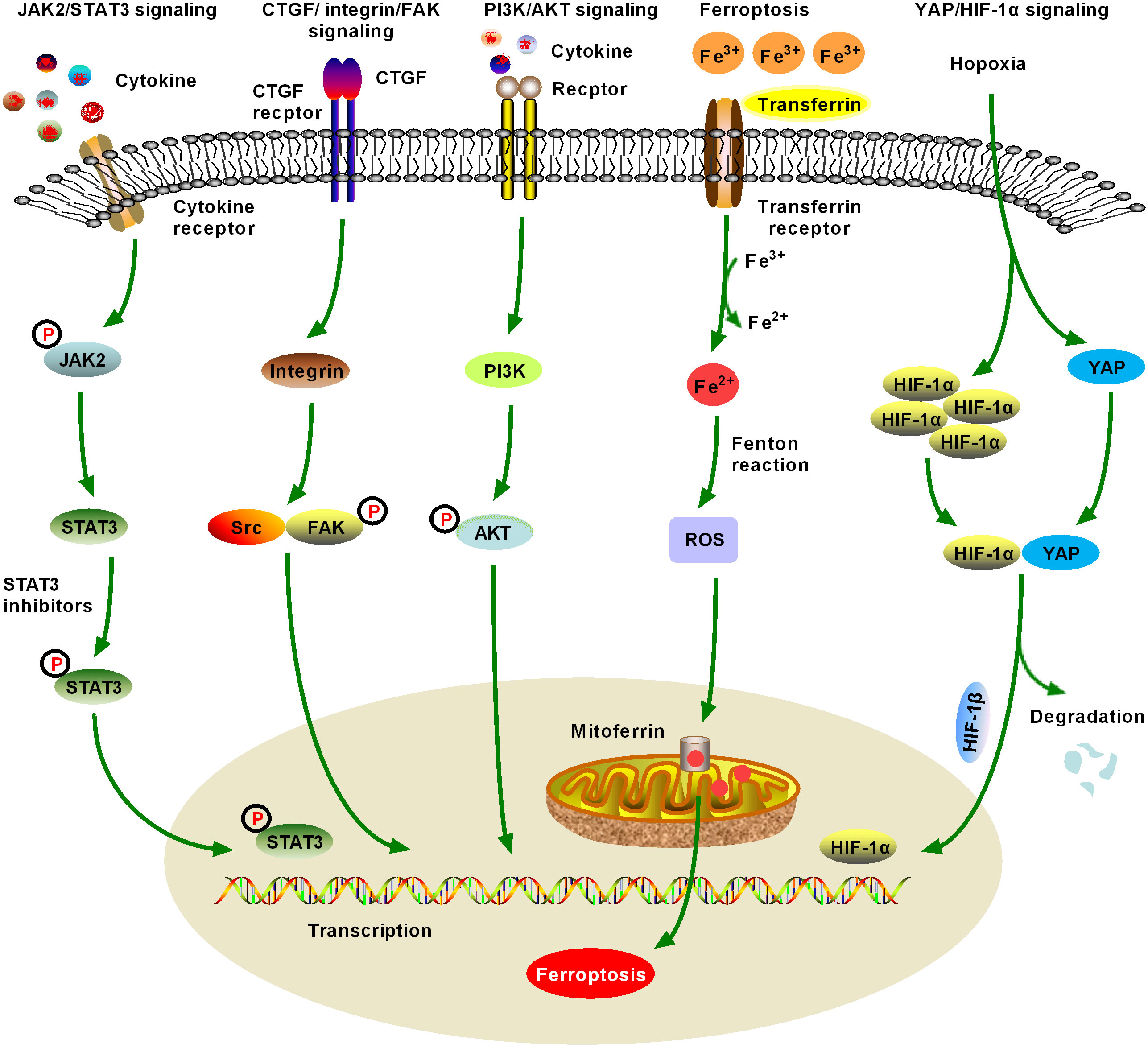
2 Fibrotic signaling in HCCThe HCC TME comprises matrix cells, including Kupffer cells, HSCs, cancer-associated fibroblasts (CAFs), liver sinusoids endothelial cells (LSECs), tumor-associated macrophages (TAMs), and lymphocytes. The HCC–HSC dialog plays an important role in the development of HCC (17). HSCs play a central role in the occurrence and development of ICC, especially in the cytokine dialog between ICC and matrix cells. Metastatic cancer cells enter the sinusoidal spaces of the liver, where most are captured and killed by Kupffer and NK cells (18). Escaped cancer cells form micrometastases and induce a microenvironment conducive to metastasis. These cells are activated following liver damage, after which, they differentiate into myofibroblast (MYF)-like cells and produce a large amount of cytokines, chemokines, growth factors, and extracellular matrix (ECM). In addition to producing and secreting collagen and other scar tissue, HSCs have other important functions, including participating in liver regeneration, immune regulation, immune tolerance, and liver tumorigenesis. Various cytokines mediate the development of HCC through multiple fibrosis signaling pathways (19). Therefore, here we review recent advances in the field of hepatic fibrosis and HCC and consider several molecular signaling pathways that contribute to HCC in the hepatic fibrosis microenvironment (20) (Figures 2, 3, Table 1).
Figure 2 Schematic of five significant fibrotic signaling pathways in HCC, including toll-like receptor 4 (TLR4)-myeloid differentiation primary response gene 88 (MyD88)-nuclear factor kappa B (NFκB) signaling, Hedgehog (Hg) signaling, MAPK/extracellular signal-regulated kinase (MEK)/extracellular regulated protein kinase (ERK) signaling, transforming growth factor-β (TGF-β) signaling, and Wnt/β-catenin signaling (1). TLR4-MyD88-NFκB signaling is one of the most important inflammatory and fibrotic pathways discovered in recent years, the activation of which releases downstream inflammatory cytokines, inducing the production of interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α. Activation of NFκB downstream of the pathway is induced, and the signal enters the nucleus to induce activation, proliferation, invasion, and migration of HCC cells, and inhibit their apoptosis (2). Hg signaling is activated in different tumors and may contribute to the development of multiple tumor types by promoting the process of tumor initiation and metastasis. Novel Hg signaling inhibitors have entered the clinical research stage for the treatment of HCC; however, the development of Hg signaling targeted inhibitors still has broad prospects (3). MEK/ERK signaling is the most active research area in cell signal transduction recently. MEK/ERK signaling transfers a variety of extracellular signals to the nucleus through phosphorylation and activates various transcription factors, regulating cell proliferation, growth inhibition, differentiation, and apoptosis. MEK/ERK signaling is an important fibrosis signal in HCC (4). TGF-β1 is an important cytokine in the development of HCC, and it is also the strongest known fibrogenic factor. TGF-β1 signaling regulates the growth and proliferation of HCC cells. Currently, TGF-β1 is highly expressed in patients with HCC, where it is significantly correlated with the degree of tumor differentiation. The expression level of TGF-β1 increased with the decrease in tumor cell differentiation, suggesting that TGF-β1 can be used as an indicator for the early diagnosis of HCC. TGF-β signaling is an important fibrosis signaling of HCC (5). After activation of Wnt/β-catenin signaling, β-catenin accumulates continuously in the cytoplasm, which promotes part of β-catenin to enter the nucleus, activate and bind to the T cell factor/lymphoid enhancer transcription factor family, initiate the transcription of multiple downstream target genes, and promote the development of HCC. Activation of Wnt/β-catenin signaling promotes the activation and proliferation of HSCs and HCC cells and is an important fibrotic signaling pathway in HCC.
Figure 3 Schematic figure of another five significant fibrotic signaling pathways in HCC, including Janus kinase2 (JAK2)/signal transducer and activator of transcription 3 (STAT3), connective tissue growth factor (CTGF)/integrin/focal adhesion kinase (FAK), and Yes-associated protein (YAP)/hypoxia-inducible factor-1α (HIF-1α) signaling (6). JAK2/STAT3 signaling can be activated by various cytokines and growth factors, resulting in dysregulation of downstream target genes, which promote the formation and metastasis of malignant tumors by controlling cell proliferation, angiogenesis, immune surveillance, tumor invasion, and metastasis. JAK/STAT3 signaling is one of the important fibrotic signaling pathways in HCC (7). Increased expression and distribution of integrins have been observed in almost all human cancers, and multiple integrin-related genes are upregulated in HCC. Currently, crosstalk between CTGF/integrin/FAK and other signaling factors, such as TGF-β, has been extensively studied (8). Numerous studies have confirmed that the activation of phosphatidylinositol 3-kinase (PI3K)/AKT signaling promotes the proliferation, migration, and glycolysis of HCC cells, and is a widely studied fibrosis signal in the context of HCC. An increasing number of PI3K/AKT signaling-targeted inhibitors have been identified for preclinical or clinical trials (9). Ferroptosis was first proposed in 2012 and has quickly become a hot research topic. Various natural products improve hepatic fibrosis by inducing ferroptosis of HSCs and myofibroblasts and prevent HCC by inducing ferroptosis in HCC cells. However, as these studies are still in the initial stage and many research results are controversial, further studies are necessary (10). Hypoxia-induced YAP/HIF-1α signaling activation facilitates tumor cell growth, survival, and metastasis, and hypoxia is a central marker of HCC and its microenvironment. YAP/HIF-1α signaling is a widely studied fibrotic signaling in HCC. HIF-1α inhibitors may be developed as HCC therapeutic drugs in the future.
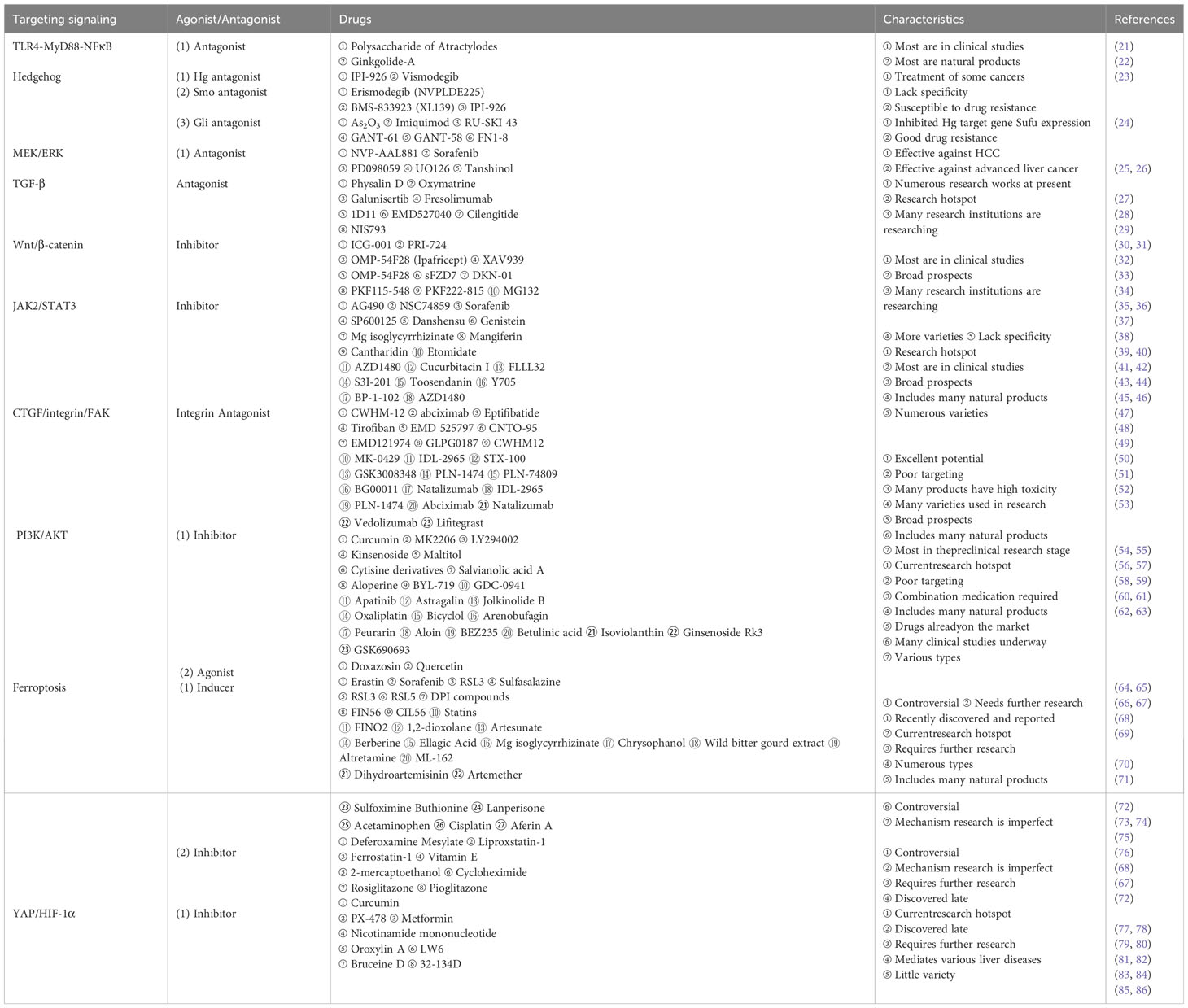
Table 1 Major drugs targeting the 10 signaling pathways.
2.1 TLR4-MyD88-NFκB signaling pathwaysActivation of the TLR4-myeloid differentiation primary response gene 88 (MyD88)-NFκB signaling pathway, an important pathway associated with the inflammatory response and hepatitis/hepatitis fibrosis, can lead to the release of downstream inflammatory factors and induce the production of interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α. TLR4 has been shown to pass through the Toll/IL-1 receptor domain (TIR domain) and use the TIR domain adaptor proteins MyD88, IL-1 receptor-associated kinase, and TNF receptor-s-sociated factor 6, to induce downstream NFκB activation to produce inflammatory factors (87). Extracellular inflammatory signals are presented to MyD88 via the TLR4-mediated signaling pathway, which activates NFκB, inducing its nuclear translocation and subsequent signal transduction (88) (Figure 2).
2.1.1 TLR4-MyD88-NFκB signaling and hepatic fibrosisHepatic fibrosis is the result of long-term chronic liver injury and repeated scar repair, which is mainly characterized by excessive deposition of ECM. HSCs are the main source of scar tissue in hepatic fibrosis. HSCs possess a complete TLR4 signaling pathway, which mediates important biological characteristics, including inflammation phenotype tolerance to apoptosis in fibrosis. Indeed, the degree of fibrosis in mice with TLR4 mutation has been shown to be significantly reduced in the three hepatic fibrosis models of bile duct ligation (BDL), carbon tetrachloride (CCl4), and thioacetamide (TAA), indicating that the TLR4 signaling pathway is involved in the occurrence of hepatic fibrosis (89, 90). Moreover, lipopolysaccharide (LPS) has been shown to cause swelling of hepatocytes, disappearance of the hepatic cord structure, and infiltration of numerous inflammatory cells. LPS has also been shown to upregulate the messenger ribonucleic acid (mRNA) and protein expression levels of TLR4, MyD88, IκBα, and NFκB, and increase the secretion of cytokines, including IL-1β, IL-4, IL-6, and TNF-α levels. Guo et al. (21) found that pretreatment with polysaccharide of Atractylodes macrocephala Koidz (PAMK) relieved LPS-induced histopathological damage in mice, and could activate TLR4-MyD88-NFκB signaling, reduce the levels of IL-1β, IL-6, and TNF-α, increase IL-4 levels, and inhibit the levels of glutathione peroxidase and malondialdehyde. These results indicate that PAMK could reduce inflammatory damage and oxidative stress in mice and play a protective role in the early stages of LPS invasion of the liver. However, overexpression of the TLR4/MyD88/NFκB axis and its downstream pro-inflammatory mediators, such as TNF-α, IL-6, and interferon (IFN)-γ, were observed in mice with CCl4-induced cirrhosis. Inhibiting the TLR4/MyD88/NFκB signaling pathway has protective effects on liver injury induced by various inflammatory cytokines (22). Activation of the TLR4-myD88-NFκB signaling pathway, an important pathway associated with hepatic fibrosis and HCC, can lead to the release of downstream inflammatory factors and induce the production of IL-1, IL-6, and TNF-α. These results suggest that inhibition of the TLR4-MyD88-NFκB-mediated inflammatory response can down-regulate the expression of TLR4 mRNA and protein, thereby improving hepatic fibrosis (91) (Table 1).
2.1.2 TLR4-MyD88-NFκB signaling pathway and HCCAt present, there are at least two views on the mechanism by which the TLR4-mediated signaling pathway induces HCC formation. The TLR4-MyD88-dependent signaling pathway can activate transcription factors such as NFκB and activatorprotein-1 (AP-1), before activating related immune cells to play the role of immunosuppression and promote the development of tumor disease (92). It has been reported that intraperitoneal injection of diethylnitrosamine (DEN) to induce HCC in animals activates the TLR4-mediated MyD88 signaling pathway, resulting in the activation of Kupffer cells, the production of IL-6 and other pro-inflammatory mediators, and the induction of carcinogenic effects of the TLR4-MyD88-dependent pathway (93). Therefore, the activation of the TLR4-MyD88 signal is considered to be one of the important causes of HCC. In-depth studies on HCC have shown that the downstream multifunctional NFκB signaling pathway regulated by TLR4 plays a key role in the induction of tumor formation by inflammatory mediators. TLR4-MyD88-NFκB signaling plays a positive regulatory role in the inflammatory progression of HCC, suggesting that the TLR4-MyD88-NFκB signaling pathway may be a new target for the prevention or treatment of HCC (94). The regeneration of hepatocytes caused by inflammatory damage induced by the activation of TLR4-MyD88-NFκB signaling pathway mostly originates from the activation of HSCs, which lead to various biological changes, including promoting the proliferation and migration of fibrous tissue and secreting several cytokines with antiapoptotic effects and the ability to promote proliferation. Chronic inflammation leads to abnormal cell growth, gene expression disorders, and DNA damage in liver tissues, which further results in malignant tissue development and ultimately induces HCC.
2.2 Hedgehog signaling pathwayThe discovery of the Hedgehog (Hg) signaling pathway stems from studies of embryonic development in Drosophila melanogaster. In humans, the pathway transmission process can be simply summarized as the Hg Homo sapiens patched 1-Smoothened-Glioma associated oncogene homolog (Ptch1–Smo–Gli) process. As the most important nuclear transcription factor in the Hg pathway, Gli-l is responsible for regulating many downstream effector factors of the pathway and binds to the promoter of downstream genes in the Hg signaling pathway to directly regulate the transcription and expression of target genes (Figure 2). Recent studies have shown that the Hg pathway is abnormally activated in various liver diseases (95, 96). In addition, the Hg signaling pathway can promote the proliferation of tumor cells, inhibit apoptosis, and promote the occurrence of HCC. Therefore, the Hg signaling pathway is closely related to hepatic fibrosis and HCC (97). The Hg signaling pathway not only participates in cell growth and differentiation but also plays an important role in tissue and organ damage repair and immune regulation. However, abnormal activation of the Hg signaling pathway can also lead to abnormal development and even tumors.
2.2.1 Hg signaling and hepatic fibrosisMany studies have shown that the immune cell-mediated microenvironment of hepatic fibrosis is closely related to the activation of the Hg signaling pathway (98). Natural killer T (NKT) cells also produce Sonic Hedgehog (SHH), promote collagen secretion, and transform stationary HSCs into MYFs, resulting in hepatic fibrosis (99, 100). The human Hg protein is highly expressed in stationary HSCs, and the key gene of the Hg signaling pathway (Gli-1 Gli-2) cannot be detected. However, after 24 h of culture, the expression of human Hg interacting protein in HSCs decreased by 90% and the Hg signaling pathway ligands SHH and Gli-1 increased significantly, leading to activation of the Hg signaling pathway (101). The pathological characteristics of hepatic fibrosis include excessive synthesis and insufficient degradation of ECM, leading to its deposition in the liver. Persistent liver fibrosis may develop into cirrhosis and increase the risk of HCC. Inhibition of the Hg pathway (using Smo antagonists or by knocking out the Smo) can reduce the activation of quiescent HSCs, reduce the production of MYF HSCs, and reduce the degree of hepatic fibrosis (Table 1). Although the Hg signaling pathway is associated with quiescent HSC activation, the activation mechanism is unclear. Although the Nobel Prize winners Wieschaus and Nussland-Volhart reported the Hg pathway in 1980, its importance in dictating hepatic fibrosis and HCC outcomes has emerged much more recently (102). Fan et al. found that suppressing the activation of HSCs could alleviate hepatic fibrosis in mice induced by CCl4 and a 3,5-diethoxycarbonyl-1,4-dihydrocollidine diet for 8 weeks via the Hg pathway (103). In mice or patients with hepatic fibrosis, inhibition of glutaminase blocked the accumulation of MYFs and fibrosis progression. Du et al. reported that knockout of the Hg signaling intermediate heptahelical transmembrane G protein-coupled receptor SMO or knockdown of Yes-associated protein (YAP) inhibited the expression of glutaminase, the rate-limiting enzyme in glutaminolysis, in mice and patients with hepatic fibrosis (99). Hg signaling regulates glutaminolysis to inhibit HSC activation. In vivo and in vitro studies confirmed that procyanidin B2, a flavonoid extract that is abundant in grape seeds and has pharmacological effects, alleviated CCl4-induced hepatic fibrosis in mice by inhibiting the activation of HSCs and angiogenesis via the Hg pathway during hepatic fibrosis (98). In other words, the Hg pathway plays an important role during various liver injuries, such as hepatic fibrosis, inflammation-related injury, and liver carcinogenesis. Targeting the Hg pathway has become a promising treatment for hepatic fibrosis.
2.2.2 Hg signaling pathway and HCCAbnormal activation of the Hg signaling pathway is closely related to the invasion and metastasis of malignant tumors. Aberrant activation of Hg signaling in the normal liver and HCC has been demonstrated in detail in previous studies (23). A new study found that the stage of hepatic fibrosis was associated with the degree of Hg signaling activation in patients with non-alcoholic fatty liver disease (NAFLD). Activation of Hg signaling is also associated with fibrosis in the lungs, skin, and kidneys (104). Chung et al. further investigated up-regulated hepatic expression of SHH-induced hepatic fibrosis and hepatocarcinogenesis in a transgenic mouse model (105). GANT61 (NSC136476) is a Gli-1- and Gli-2-induced transcriptional inhibitor that inhibits Hg. Wang et al. found that GANT61 significantly suppressed Hg signaling to reverse sorafenib resistance in CD44-positive HCC tissues (97). This combined administration may be effective in patients with HCC with high CD44 levels as a personalized medicine approach. Another study has shown that the Hg pathway in liver tissue of Chinese patients with HCC is activated by ligand expression, rather than by mutations, suggesting that the research prospects of Hg signaling inhibitors are very broad for a large number of patients with HCC in China (24). Currently, the role of the Hg signaling pathway in liver physiology and pathology has not been fully elucidated and requires further study.
2.3 MEK/ERK signaling pathwayHSC activation is facilitated by several mitogen-activated protein kinases (MAPKs), including MAPK/extracellular signal-regulated kinase (MEK), extracellular regulated protein kinase (ERK), connective tissue growth factor (CTGF), and insulin-like growth factor-1 (106). MEK binding to ERK results in dimerization of tyrosine (Tyr) residues, which activates the phosphatidylinositol 3-kinase (PI3K) and MAPK pathways. Blocking MEK/ERK signaling blocks MAPK and PI3K/AKT signaling pathways, thereby inhibiting HSC activation and attenuating experimental hepatic fibrosis progression (Table 1). ERK/MAPK signaling pathways are activated and involved in cell growth, differentiation, and migration during the pathogenesis of hepatic fibrosis, cirrhosis, and HCC (25, 107). The MEK/ERK signaling pathway is closely related to hepatic fibrosis and HCC (Figure 2).
2.3.1 MEK/ERK signaling and hepatic fibrosisStudies have confirmed that ERK is a major member of the MAPK family and that its MEK/ERK signaling pathway is an important pathway for many cytokines to regulate cell proliferation and apoptosis. Foglia et al. suggested that inhibiting the MEK/ERK pathway in activated MYF-like HSCs is a key crossroad for reversing hepatic fibrosis (26). HSC activation by myofibroblastic differentiation is critical for hepatic fibrosis. Crosstalk between HSCs/myofibroblastic and tumor cells in the microenvironment alters the properties and facilitates the growth, proliferation, migration, and invasion of HCC cells. Homo sapiens E2F transcription factor 3 (E2F3) acts as a transcriptional activator and increases cell proliferation through G1/S transformation. A new report has identified a novel stiffness-mediated HSC activation mechanism that is dependent on the E2F3 (108). Liu et al. found that HCC cells cultured in an HSC-conditioned medium activated the PI3K/AKT and MEK/ERK signaling pathways following the combination of E2F3 with the fibroblast growth factor 2 (FGF2) promoter, which increased the growth and metastasis of HCC cells. In addition, gene knockout of E2F3 mitigated DEN- and CCl4-induced HCC in mice. The E2F3 is also highly expressed in the HCC tissues of patients; therefore, matrix stiffness modulates HSC activation into tumor-promoting MYFs via E2F3-dependent MEK/ERK signaling and regulates malignant progression. The ERK pathway is critical for transducing signals from surface receptors to the nucleus and is overactivated in many tumors, including HCC, melanoma, and breast cancer.
2.3.2 MEK/ERK signaling and HCCAccording to new statistics, the majority (approximately 80%) of HCC cases result from severe hepatic fibrosis and/or cirrhosis. Li et al. found that the serum cartilage oligomeric matrix protein (COMP) levels in patients with HCC were obviously higher than those in healthy people, and these patients showed more unfavorable disease parameters, including a higher incidence of vascular invasion and HCC recurrence (106). In addition, gene knockout animal experiments and different cell experiments demonstrated for the first time that COMP mainly originates from activated HSCs and promotes the growth and metastasis of HCC cells in a dose-dependent manner by activating MEK/ERK and PI3K/AKT signaling. Moreover, crosstalk was observed between hepatic fibrosis and HCC through inhibiting ERK signaling, which is a potential novel target for the prevention and treatment of HCC. Another study showed that low-density lipoprotein receptor inhibited the enhancement of intracellular cholesterol synthesis through MEK/ERK signaling and promoted the proliferation and metastasis of HCC cells (109). It has also been reported that Homo sapiens minichromosome maintenance complex component 6 promotes HCC metastasis through MEK/ERK signaling and can be used as a novel serum biomarker for early recurrence (110). Lai et al. found that NEI-like DNA glycosylase III (NEIL3) activation of MEK/ERK signaling mediated epithelial–mesenchymal transition (EMT), that treatment resistance promoted HCC progression, and that NEIL3 induction of targeted inhibition of NEIL3 was a promising therapeutic approach in patients with HCC (111). Currently, sophoridine derived from natural products has been found to inhibit the growth of lenvatinib-resistant HCC by inhibiting rat sarcoma virus (RAS)/MEK/ERK signaling by decreasing vascular endothelial growth factor receptor 2 expression (25). Another natural product, Morusinol, inhibits HCC cell invasion and migration and targets RAS/MEK/ERK signaling by inducing autophagy, and has selective and effective antitumor activity against human HCC (112). These studies further confirm that inhibition of MEK/ERK signaling alleviates HCC.
2.4 TGF-β signaling pathwayDiscovered by Tucker in 1984, transforming growth factor-β (TGF-β) is a polypeptide cell growth regulatory factor associated with the growth of various tumors. The TGF-β superfamily members include at least five isomers (TGF-β1, 2, 3, 4, 5), among which TGF-β1 is the most closely related to liver injury and disease occurrence (113). TGF-β1 is a secretory polypeptide factor, which has various biological activities, including regulating cell growth, migration, differentiation, the occurrence and development of embryos and tumors, wound healing, bone formation, and immune regulation. Normal hepatocytes either lack TGF-β1 or show low levels (114). Meanwhile, damaged liver endothelial cells cause platelets to agglutinate and release TGF-β1. Studies have shown that TGF-β1 is a crucial factor leading to hepatic fibrosis and even HCC in the process of liver injury. The drosophila mothers against decapentaplegic protein (Smad) is the substrate of the most important intracellular kinase of the TGF-β1 receptor known to date. Activated TGF-β1 receptors recruit Smads through Smad anchor proteins or directly bind to signal molecules such as MAPK and phosphorylate them, so that signals are transmitted step by step in the cell until they are transferred to the nucleus, where they regulate the expression of target genes (115) (Figure 2).
2.4.1 TGF-β signaling and hepatic fibrosisMost literature studies have shown that TGF-β1 stimulates HSC activation and proliferation, leading to hepatic fibrosis (27, 28, 116, 117) (Table 1). TGF-β1 in the liver is mainly secreted by immune cells, HSCs, and epithelial cells, mainly through mediating the activation of HSCs to produce excessive ECM, leading to hepatic fibrosis (117). Xiang et al. reported that Physalin D alleviated CCl4- and BDL-induced hepatic fibrosis in mice via blocking TGF-β/Smad signaling and reducing HSC activation, proliferation, and transformation (27). Compound kushen injection, an approved traditional Chinese medicine formula, reduced the inflammatory response, oxidative stress, liver compensatory proliferation, and hepatocellular death of mice with hepatic fibrosis induced by CCl4 injection or a methionine choline-deficient diet via rebalancing TGF-β/Smad7 signaling in HSCs, which protected against hepatic fibrosis and hepatocarcinogenesis in both preclinical and clinical studies (28). Sulfatase-2 (Sulf2) also regulates hepatic fibrosis in mice induced by BDL or intraperitoneal injection of CCl4 or TAA through inhibiting TGF-β signaling (118). Sulf2 knockout (Sulf2-KO) mice showed significantly decreased collagen content and bands of bridging fibrosis compared with wild-type mice in all three models of hepatic fibrosis. Sulf2 expression has recently been shown to be upregulated in cirrhotic human liver and fibrotic mouse liver, suggesting that Sulf2 plays an important role in both fibrosis and subsequent tumorigenesis. TGF-β is highly expressed in tissues of hepatic fibrosis and HCC, so the TGF-β signaling is considered to be a marker of hepatic fibrosis and HCC. The synergistic role of TGF-β and the tissue microenvironment in modulating the cellular response of different cell types and promoting the development of hepatic fibrosis and the progression of HCC has been extensively demonstrated (29). TGF-β signaling provides a wide scope for intracellular crosstalk, such as receptor-associated Smads interacting with other signaling molecules rather than directly transmitting signals to the nucleus, and the activation of intracellular substrates other than Smad can be mediated by influencing apoptosis and other activation of other intracellular signaling pathways (119). The mechanism of treating hepatic fibrosis through targeted inhibition of TGF-β signaling has been widely discussed (119, 120).
2.4.2 TGF-β signaling and HCCTGF-β1 is an important cytokine in the occurrence and development of HCC. As well as being an important cytokine that controls the growth and proliferation of hepatocytes, TGF-β1 is also known to be highly expressed in patients with HCC and is significantly correlated with the degree of tumor differentiation (29). Recently, cancer-associated fibroblast-mediated cell crosstalk supporting HCC progression has become a research hotspot (121). CAFs are key players in the pathogenesis of HCC; however, the complex mechanism of crosstalk between CAFs and other components of the TME is still unclear, and studies on TGF-β1 activation of CAFs are ongoing (121). Aberrant activation of TGF-β/Smad signaling facilitates tumor metastasis and is often observed in HCC. The lncRNAlnc-UTGF has been shown to mediate a positive feedback loop regulates TGF-β/Smad signaling and promotes hepatoma metastasis (122). Moreover, bone morphogenetic protein (BMP) is involved in TGF-β signaling crosstalk; indeed, Ning et al. found that TGF-β1/BMP-7 pathway imbalance induced by activation of liver polarized macrophages promotes the aggressiveness of HCC (123). Increasing evidence has shown that TGF-β signaling plays a critical role in the regulation of immune cells, including CD4+ T cells, CD8+ T cells, dendritic cells (DCs), NK cells, myeloid suppressor cells, and TAMs. TGF-β signaling mediates HCC progression through the critical regulation of various immune cells in the liver to maintain a balance between immune tolerance and activation (124). RALYL, a liver progenitor-specific gene, was recently found to promote tumorigenicity, self-renewal, chemotherapy resistance, and metastasis of HCC by up-regulating TGF-β signaling and subsequent PI3K/AKT and signal transducer and activator of transcription 3 (STAT3) signaling to enhance HCC stemness (125). Increasing evidence suggests that crosstalk between ECM and TGF-β, and intracellular TGF-β signaling often exhibits crosstalk with other signals, including Jun N-terminal kinase, p38, MAPK, and NFκB, to regulate hepatic fibrosis and HCC (126). Some studies have explored the underlying molecular mechanisms of STAT3 crosstalk with Smad3/TGF-β1 signaling during EMT in patients with HCC and a rat model. Both in vivo (HCC patients, DEN-induced HCC rat model) and in vitro (HepG2, Bel7402, MHCC97H, and HCCLM3) results indicate that TGF-β1-induced EMT is dependent on Smad3-mediated Snail transcription and crosstalk of STAT3 signals in HCC cells (30). Interestingly, new studies have shown that TGF-β has a dual role in tumorigenesis, acting as a tumor suppressor in the early stages of tumorigenesis and promoting tumor progression and metastasis in more advanced cancers (31). In addition, the crosstalk between TGF-β and vascular endothelial-derived growth factor (VEGF) signals in a variety of immune cells, tumor cells, and matrix cells may further promote TGF-β-mediated immunosuppression, which may be a new mechanism for TGF-β to regulate tumor immune escape through immunosuppression in the latest tumor stage (126). However, TGF-β1 plays a role in promoting tumor growth and metastasis in the middle and late stages of the tumor. Overexpression of TGF-β1 in HCC cells does not inhibit the proliferation of HCC cells, which may be related to the defective receptor.
2.5 Wnt/β-catenin signalingWnt/β-catenin signaling comprises a group of evolutionarily conserved signals that play an important role in cell genesis, embryonic development, cell proliferation and differentiation, and tissue regeneration in various organisms (35). Recent studies have shown that Wnt signaling can participate in the activation of HSCs and the formation of hepatic fibrosis, which is mainly manifested in promoting collagen synthesis and collagen deposition in the liver by means of autocrine and/or paracrine mechanisms, thus reducing the apoptotic capacity of activated HSCs (36) (Table 1). Various stimulators can also play a role through the classical Wnt/β-catenin signaling pathway, ultimately promoting the occurrence and development of hepatic fibrosis. Other studies have found that abnormal activation of Wnt/β-catenin signaling is closely related to the progression and carcinogenicity of malignant tumors (127) (Figure 2). Specifically, the expression level of intracellular β-catenin is closely related to tumor metastasis and invasion of blood vessels and may lead to tumor enlargement. Wnt/β-catenin signaling is closely related to hepatic fibrosis and HCC.
2.5.1 Wnt/β-catenin signaling and hepatic fibrosisActivation of classical Wnt/β-catenin signaling can regulate cell proliferation, survival, and behavior. New studies have found that a series of intracellular signals drive HSC activation, including Wnt/β-catenin signaling, TGF-β/Smad signaling, and Hg signaling, with complex crosstalk between them (32). Increasing evidence suggests that inhibition of Wnt signaling to β-catenin may alleviate hepatic fibrosis; however, further research is needed to determine the identity and cellular origin of the factors that activate β-catenin in HSCs (32). Liu et al. found that insulin-like growth factor binding prote-3, Dickkopf-3 (DKK-3), and DKK-1 derived from human amniotic mesenchymal stem cells (MSCs) mitigate hepatic fibrosis via suppression HSC activation through blocking Wnt/β-catenin signaling in mice induced by injecting CCl4 via the tail vein. In addition, crosstalk between PI3K/AKT and Wnt/β-catenin signals were found during HSCs (LX-2) transfection assays in underlying mechanism studies (128). Rong et al. found that human bone marrow MSC-derived exosomes reduced CCl4-induced hepatic fibrosis in mice through Wnt/β-catenin signaling via both cellular assays and animal experiments (129). Upstream regulatory mechanisms that inhibit hepatic Wnt/β-catenin activity may constitute targets for the development of new therapies against life-threatening diseases such as hepatic fibrosis and HCC (130). Therefore, the regulation of β-catenin is expected to become a target of anti-hepatic fibrosis therapy, as well as a direction of future research.
2.5.2 Wnt/β-catenin signaling and HCCNumerous literature studies have shown that inhibition of Wnt/β-catenin signaling can inhibit HCC cell differentiation and proliferation and promote apoptosis during HCC progression (33, 131, 132). TAMs are an important component of the TME mediating the development of HCC. Yang et al. confirmed for the first time that the typical Wnt/β-catenin signaling crosstalk between HCC cells and macrophages promotes the polarization of M2-like macrophages, thus leading to tumor growth, migration, metastasis, and immunosuppression in HCC (133). Therefore, blocking Wnt/β-catenin signaling activation in HCC cells secreting Wnt and/or TAMs may be a potential strategy for future HCC treatment. Up to 70% of patients with HCC show up-regulation of Wnt/β-catenin signaling, and β-catenin mutations increase with HCC progression (134). Wang et al. reported that brain-expressed X-linked protein 1 (BEX1) plays a critical role in regulating cancer stem cell (CSC) properties in different types of HCC; indeed, targeting BEX1-mediated Wnt/β-catenin signaling may help to address the high recurrence rate and heterogeneity of HCC (135). Lentivirus-mediated overexpression or CRISPR/Cas9 knockout experiments have shown that glutaminase 1 (GLS1) regulates stemness and serves as a therapeutic target for the elimination of CSCs by inhibiting reactive oxygen species (ROS)/Wnt/β-catenin signaling in vitro and in vivo, while GLS1 knockout inhibits tumorigenicity in vivo (136). Protein kinases play a key evolutionary conserved role in Wnt/β-catenin signaling and have been widely discussed as one of the most important drug targets in HCC therapy (34). However, inhibition of Wnt/β-catenin signaling alone is unlikely to significantly improve the prognosis of patients with HCC, and future research should focus on the combination of other therapies to improve the efficacy of Wnt/β-catenin signaling inhibitors. Currently, anti-HCC compounds targeting the inhibition of Wnt/β-catenin signaling are the most widely researched, most of which are in the clinical and preclinical research stages. As more research data become available, achieving the best personalized treatment for HCC in the future represents the ideal goal (137–140). Inhibition of Wnt/β-catenin signaling reduces the activation, proliferation, migration, and progression of HCC cells, which has attracted extensive attention from many researchers worldwide. At present, several institutions are developing targeted drugs.
2.6 JAK2/STAT3 signalingMany studies suggest that Janus kinase2 (JAK2)/STAT3 signaling is closely related to hepatic fibrosis, and HSC activation is a key factor in the progression of hepatic fibrosis (39). Following the occurrence of homologous or heterooligomerization of specific receptor subunits on the HSC membrane with platelet-derived growth factor, leptin, TGF-β, and other cytokines promoting hepatic fibrosis, the receptor-coupled JAKs are activated. The Tyr of the cytoplasmic segment of the receptor is then phosphorylated to the protein anchor site containing SH2, and downstream signaling protein molecules are activated to enable the target genes to play a role. Tyr residues (Tyr705) and Serine (Ser) residues (Ser727) at the end of the STAT protein can also be phosphorylated by JAKs to activate the STAT protein and form homologous or heterologous STAT protein dimers into the nucleus to bind the promoter of corresponding target genes, thus activating target gene transcription of HSCs and participating in the development of hepatic fibrosis (38). The results of animal experiments have shown that p-JAK2/JAK2 and p-STAT3/STAT3 protein levels were significantly higher in the fibrosis model group compared with those in the control group, suggesting that JAK2/STAT3 signaling was activated in the liver tissues of rats during dimethylnitrosamine-induced hepatic fibrosis and was involved in the development of hepatic fibrosis (40) (Figure 2). Moreover, many studies have shown that JAK2/STAT3 signaling activation is involved in the occurrence and development of hepatic fibrosis and HCC (Table 1).
2.6.1 JAK2/STAT3 signaling and hepatic fibrosisJAK2/STAT3 signaling is a research hotspot in fibrotic disease, but the role of JAK2/STAT3 in the progression of hepatic fibrosis remains controversial (37, 141). Ding et al. found that HCC is closely related to chronic inflammation and fibrosis, which is called the inflammation–fibrosis–cancer axis (142). In addition, compared with the control group, JAK2/STAT3 signaling was significantly upregulated in the DEN exposure group, in that 100% of the rats in the DEN exposure group developed liver tumors at 20 weeks, with accompanying inflammatory and fibrotic stages correlated with exposure time. Most literature studies have reported that inhibiting JAK2/STAT3 signaling of HSC alleviates hepatic fibrosis both in vivo and in vitro (38, 39). Yang et al. found that magnesium (Mg) isoglycyrrhizinate improved high-fructose-induced liver fibrosis in rats by inhibiting JAK2/STAT3 and TGF-β1/Smad signaling by increasing microRNA-375-3p, but the effect of a direct interaction between JAK2/STAT3 and TGF-β1/Smads signaling on hepatic fibrosis in vivo needs further study (143). Studies have confirmed that quiescent HSCs, hepatocytes, and bile duct cells with an epithelial phenotype can be transformed into fibroblasts with a mesenchymal phenotype through EMT and participate in the occurrence and development of hepatic fibrosis (40). Xu et al. reported that the natural extract of brown algae, named propylene glycol alginate sodium sulfate (PSS), could significantly prevent liver injury and fibrosis induced by BDL and CCl4 in mice. Mechanistically, PSS significantly inhibits the activation of HSCs in LX-2 cell lines incubated with 10 ng/mL TGF-β1 for 24 h by inhibiting the anti-autophagy effect of JAK2/STAT3 signaling (144). In conclusion, STAT3 may play a dual role in hepatic fibrosis because of different upstream regulatory factors. Currently, most studies suggest that STAT3 mainly plays a role in promoting hepatic fibrosis, and its expression level may be positively correlated with the degree of fibrosis.
2.6.2 JAK2/STAT3 signaling and HCCSTAT3 is closely related to the development and prognosis of HCC and can be adjusted by different target genes, affecting cell proliferation, apoptosis, invasion, migration, angiogenesis, and immune escape. Many studies have reported that directly inducing apoptosis of HepG2 cells by downregulating the JAK2/STAT3 signal transduction pathway may contribute to the development of new therapeutic strategies for HCC (145). The humanized-immune-system HCC mouse model is a newly developed animal model for the study of new targets of HCC immunotherapy, which has wide application prospects (146). Zhao et al. first demonstrated that intratumor human cluster of differentiation–positive (hCD14+) cells could produce IL-33 through DAMP/TLR4/activator protein 1, which increased IL-6 in other intratumor immune cells and activated JAK2/STAT3 signaling in HCC (146). However, the mechanism of crosstalk between human HCC and the human immune system still needs to be studied in depth (41, 147). Wei et al. recently found that TAMs mediate the EMT program of tumor cells before the invasion and speculated that EMT-programmed tumor cells can in turn recruit macrophages. In addition, crosstalk exists between TAMs and cancer cells in the TME, and IL-6 secreted by TAMs binds to the IL-6 receptor on the surface of cancer cells and phosphorylates STAT3 (pSTAT3) (148). It has been widely verified that abnormal activation (phosphorylation) of JAK/STAT3 signaling upregulates EMT, promoting HCC progression (42, 149). Liu et al. found that the homologous to the E6-AP carboxy terminus domain and RCC1-like domain 2 (HERC2) promote inflammation-induced stemness and immune evasion in HCC cells through JAK2/STAT3 signaling, the underlying mechanism of which is related to the regulatory effect of HERC2 on JAK2/STAT3 signaling participating in the crosstalk between cancer stemness and immune evasion (150). Cytokine activation of STAT3 is mediated by Tyr phosphorylation of JAK, and crosstalk between JAK/STAT3 signaling activation and EMT-mediated metastasis has been observed in HCC over the past decade (43–45). JAK2/STAT3 signaling is involved in many biological processes, including cell proliferation, angiogenesis, and migration in HCC (46).
2.7 CTGF/integrin/FAK signalingCTGF is a multifunctional secretory polypeptide that regulates biological activities such as cell proliferation, differentiation, and adhesion and plays an important role in tissue damage repair and ECM synthesis (151). Integrins are receptor proteins on the surface of HSCs. Recent studies have confirmed that CTGF plays a positive regulatory role in the development of hepatic fibrosis (152). Focal adhesion kinase (FAK) is an important cytokine in integrin signaling, and activation of FAK further initiates a series of downstream cell signaling cascades (Figure 3). Currently, many studies have found that CTGF/integrin/FAK signaling is associated with hepatic fibrosis and HCC (47, 48, 153) (Table 1).
2.7.1 CTGF/integrin/FAK signaling and hepatic fibrosisFAK is an important cytokine in the integrin signaling system. After the integrin binds to the extracellular ligand molecule, the cytoplasmic end binds to the N end of the FAK protein to transmit extracellular signals (Figure 3). FAK is activated by phosphorylation, which further activates a series of downstream protein kinases (152). Currently, invasive biopsy is still the gold standard for the diagnosis of hepatocellular fibrosis. Shao et al. developed a new [18F]-Alfatide imaging targeting integrin αvβ3, which provides a noninvasive method for evaluating the expression and function of integrin αvβ3 on activated HSCs in the injured liver (47). Researchers have successfully quantitatively evaluated the level of liver integrin αvβ3 and the progression of hepatic fibrosis in mouse models induced by CCl4 and BDL; however, these studies are only at the preclinical stage, and further studies are needed (48, 153, 154). HSC activation and MYF differentiation are central to hepatic fibrosis and occur during ECM deposition (49, 155, 156). Integrin has been extensively investigated as a drug target for hepatic fibrosis, and a variety of antifibrotic drugs targeting integrin have entered the clinical research stage (50, 51, 157). Currently, clinically used integrin inhibitors, such as abciximab (targeting αIIbβ3 on platelets), natalizumab (targeting α4β1 on T-cells), vedolizumab (targeting α4β7 on T-cells), and lifitegrast (targeting αLβ2 on T cells), usually bind specifically to their target (52). Shi et al. found that the promoter methylation of the CTGF underwent phenotypic changes in HSCs, becoming MYF-like cells expressing α-SMA. CTGF promoted phenotypic changes of HSCs into MYFs, whereas inhibition of CTGF promoter methylation enhanced this process, suggesting that CTGF group promoter methylation may negatively regulate hepatic fibrosis (158). In vivo experiments showed that the severity of hepatic fibrosis in a CCl4-induced rat liver fibrosis model was negatively correlated with the promoter methylation level of the CTGF in HSCs, suggesting that promoter methylation of the CTGF may prevent the occurrence of hepatic fibrosis. Therefore, low levels of promoter methylation of the CTGF may be a predisposing factor for the occurrence of hepatic fibrosis (158). After transfection of the FAK shRNA recombinant plasmid into HSCs, the migration and adhesion activities of the recombinant HSCs were significantly decreased compared with those of the control HSCs. In addition, FAK mRNA expression was significantly increased in acetaldehyde-activated HSCs, suggesting that FAK plays an important role in the activation of HSCs in hepatic fibrosis.
2.7.2 CTGF/integrin/FAK signaling and HCCHCC usually develops from hepatic fibrosis. CTGF has been found to be overexpressed in 93 human HCC tissues compared with human non-HCC tissues—mainly in HCC cells—and increased CTGF expression was associated with the clinicopathological malignancy of HCC (159). A previous study confirmed that tumor cell-derived CTGF is a building block in the HCC microenvironment, activating nearby HSCs and delivering growth-promoting signals to HCC cells (159). This interaction is easily suppressed by anti-CTGF antibodies, suggesting that pro-tumor crosstalk between HCC cells and HSCs provides an opportunity for therapeutic intervention in HCC. Li et al. found that activation of the integrin β1/Piezo1 contributes to matrix stiffness-induced angiogenesis in HCC, and that high Piezo1 expression is predictive of poor prognosis (160). Integrins are key players in the spread of tumor cells and are expressed at different levels at different stages of tumor development (161, 162). Sarker et al. have extensively summarized the effects of integrin crosstalk with growth factor receptors on growth factor signaling and reviewed the evidence supporting the mechanistic regulation of integrin crosstalk with growth factor signaling, which has important implications for normal cell physiology and anticancer therapy (163). A new study has observed that enhanced radiation-induced G2/M cell cycle arrest is dependent on crosstalk between integrinβ1 and growth factor receptor signaling, which is a new research direction to elucidate the signaling underlying the EGFR/integrinβ1 crosstalk, which may support the development of advanced molecular targeted therapies in radiation oncology (164). Integrin crosstalk refers to the mechanism by which changes in the expression of one integrin subunit or activation of an integrin heterodimer may interfere with the expression and/or activation of other integrin subunits in the same cell. This phenomenon was first described in K562 erythroleukemia cells in 1994 (53, 165, 166). Samaržija et al. reviewed the evidence for integrin crosstalk in a range of cellular systems, with a particular emphasis on cancer, and described the molecular mechanisms of integrin crosstalk, the influence of cell fate determination, and the contribution of crosstalk to therapeutic outcomes (53). As a key player in many cancer features, integrins have been recognized as valuable tumor therapeutic targets (167, 168). Gahmberg et al. conducted a detailed summary of the regulation of integrin-integrin crosstalk on dynamic cell adhesion and found that although the mechanisms leading to integrin crosstalk are incompletely understood, they usually involve intracellular signaling and are also used by other cell surface receptors (169). The phosphorylation of integrins and key intracellular molecules play a crucial role in integrin-cytoplasmic interactions, which, in turn, influence integrin activity and crosstalk, with the integrin β-chain playing a central role in regulating crosstalk. In addition to integrin-integrin crosstalk, crosstalk can occur between integrins and related receptors, including other adhesion and growth factor receptors.
2.8 PI3K/AKT signaling pathwayPI3K/AKT signaling plays a pivotal role in intracellular and extracellular signal transduction and is also involved in cell proliferation, apoptosis, invasion, and metastasis. PI3K is a crucial protein molecule involved in PI3K/AKT signaling (170). Activated PI3K can promote changes in the downstream substrate AKT and also activate the translocation of AKT to the nucleus, thus causing the expression of downstream-related genes and regulating cell proliferation, metabolism, and a series of physiological functions (Figure 3). Mammalian target of rapamycin is the downstream molecule of AKT and is normally activated to play an important role in the regulation of the cell cycle, cell growth, and proliferation (171). The mechanism of action of dexmedetomidine, apatinib, rosiglitazone, sorafenib, metformin, and baicalin may be downregulating the expression of PI3K and phosphorylation of AKT, while activating the apoptotic signaling of caspases to promote apoptosis. PI3K/AKT signaling is activated in 30%–50% of patients with HCC, and upregulation of phosphorylated AKT (p-AKT) is associated with poor survival and tumor vascular invasion in patients with HCC (172) (Table 1). PI3K/AKT signaling is also associated with the inhibition of HCC apoptosis. Therefore, PI3K/AKT signaling may be key to HCC drug development.
2.8.1 PI3K/AKT signaling and hepatic fibrosisThe PI3K/AKT signaling pathway is involved in many processes that regulate HSC activation, including collagen synthesis and cell proliferation. HSC activation is a key step in hepatic fibrosis that requires global reprogramming of gene expression, which is regulated by multiple mechanisms, including epigenetic regulation, such as DNA methylation (173, 174). Studies have confirmed that phosphoenolpyruvate carboxykinase 1 deficiency promotes platelet-derived growth factor AA expression through PI3K/AKT signaling and activates HSCs via hepatocyte-HSC crosstalk, and that this important crosstalk between hepatocytes and HSCs is mediated by paracrine signaling (54, 55). However, the effect of activating or inhibiting PI3K/AKT signaling on hepatic fibrosis remains controversial (64, 175, 176). Recent studies have shown that TGF-β crosstalk with PI3K/AKT signaling occurs in a Smad-independent manner, suggesting that specific crosstalk between macrophages and HSCs via soluble proteins could be an area for further study (177). Damaged hepatocytes and activated immune cells convert quiescent HSCs into MYFs through crosstalk. Recently, it has been reported that DC function is associated with PI3K/AKT signaling, involving functional reprogramming of immune cells, control of cellular responses, and regulation of hepatic fibrosis (56). PI3K/AKT signaling is involved in reprogramming the activity of many immune cells, particularly DCs and macrophages. The latest study by Xiang et al. showed that kinsenoside treatment improved the hepatic inflammatory microenvironment of hepatic fibrosis and reprogrammed intracellular glycolysis by inhibiting the migration and maturation of DCs via inhibition of PI3K/AKT signaling (56). PI3K/AKT signaling is an important pathway regulating HSC proliferation and transdifferentiation in the liver (57, 58). Further investigation revealed that hepatic fibrosis involved complex mutual crosstalk between PI3K/AKT signaling and TGF-β1/Smads signaling, and further study is required (59, 65).
2.8.2 PI3K/AKT signaling and HCCPI3K can also inhibit tumor cell apoptosis, enhance cell resistance to chemotherapy-induced apoptosis, and promote postoperative tumor growth (60, 178, 179). Crosstalk between stromal and HCC cells changes the characteristics of HCC cells and promotes their growth and metastasis. FGF is also known to play an important role in the development of HCC. Indeed, Liu et al. have demonstrated that matrix stiffness contributes to the mechanical signal transduction of HSCs and promotes the growth and metastasis of HCC cells through the secretion of FGF2 (108). Li et al. also reported for the first time that activated HSC-derived COMP regulates the gene expression of mesenchymal and matrix metalloproteinase (MMP) in HCC cells through CD36, causing abnormal phosphorylation of ERK and AKT, with crosstalk between PI3K/AKT and MEK/ERK signaling (106). Intriguingly, zebrafish share the same molecular pathways as humans. Experiments using a zebrafish HCC model found that aloperine reduced the proliferation of Huh7 cells in a dose- and time-dependent manner, suggesting that the PI3K/AKT cell cycle is an important central node against HCC (61, 180, 181). PI3K/AKT signaling is responsible for metabolic reprogramming and glycolytic induction in HCC and is therefore a promising therapeutic target. Targeting PI3K/AKT signaling has a positive biological impact and a very promising therapeutic prospect in the prevention of HCC (62). Many studies have reported that PI3K/AKT is widely expressed in various types of cancer cells, representing a promising target for tumor therapy (63, 182–185). Metabolic reprogramming is a novel feature of cancer that involves multiple effects and steps and has been recognized as a hallmark of cancer (186, 187). Moreover, the reprogramming of gene expression during EMT and non-transcriptional changes are triggered and regulated by signaling in response to extracellular signals (
留言 (0)