
記住我

Endometrial carcinoma (EC) is the most common gynecologic malignant epithelial tumor type in the United States, with the death rate from this disease increasing by more than 100% over the past 20 years (1, 2). Worldwide, EC is the sixth most common cancer among women (3). According to recent estimates, there were ~90,000 deaths and ~382,000 new cases of EC in 2018 (4). In the United States, >66,000 new cases and >13,000 deaths from EC were predicted for 2023. Most EC deaths occur in middle-aged or older women, and uterine cancer is the fourteenth leading cause of cancer death. The age-adjusted death rate is estimated to be 5.1 per 100,000 women per year (5). EC is a devastating and common disease that results from the uncontrolled growth of cells within the endometrium, the inner layer or mucosal lining, of the mammalian uterus, which is comprised of an epithelial layer, glands, connective tissue (stroma), and blood vessels (6). Endometrial tissue is responsive to hormones, and the most common type of EC is thought to arise from estrogen stimulation that is unopposed by progestins.
Surgical staging provides vital information to predict the course of EC (i.e., prognostic information about how the cancer will affect an individual and respond to treatment). EC tumors are assigned an International Federation of Gynecology and Obstetrics (FIGO) histological grade based on the level of glandular differentiation. Grade 1, 2, and 3 tumors exhibit ≤5%, 6 to 50%, and >50% solid non-glandular, nonsquamous (non-flat cell) growth, respectively (7). Grade 1 and 2 tumors are referred to as “low-grade” tumors (more of the cells form glands), while grade 3 tumors are called “high-grade” (more of the cells do not form glands and are disorganized). Grade 3 tumors tend to be more aggressive and spread and grow fast. Women with early-stage/low-grade tumors have a more favorable prognosis compared to those with advanced disease/high-grade tumors (8).
ECs are generally classified into two types of tumors. Type I endometrioid adenocarcinoma tumors are the most common, representing more than 80% of EC cases, and are associated with unopposed estrogen stimulation. Interestingly, estrogen-mediated reactive oxygen species (ROS) production has been proposed to be a contributor to mitochondrial DNA (mtDNA) mutation (9). Type I ECs are generally low-grade tumors that exhibit glandular differentiation and likely originate from glandular cells, e.g., grades 1 and 2 endometrioid cancers (10). Type I ECs most often occur in obese post-menopausal women (and sometimes in anovulatory pre-menopausal individuals) and are associated with down-regulation or mutation of the PTEN tumor suppressor gene leading to protein kinase B (Akt) and mTOR (mammalian target of rapamycin, a phosphatidylinositol kinase-related kinase) activation (11). Type II tumors account for 10% of ECs and are associated with 40% of related deaths (12).
Type II ECs include heterogenous, undifferentiated carcinoma, carcinosarcoma (a mixture of carcinoma and sarcoma), serous carcinoma, clear cell carcinoma, and grade 3 endometrioid carcinoma. Type II ECs are less associated with estrogen stimulation and are typically poorly differentiated or high-grade tumors (13). Type II ECs are reported to be related to abnormal TP53, HER2 (ErbB2), and P16 and are often metastatic and associated with poor survival despite aggressive treatments with radiation and chemotherapy (11). A study by The Cancer Genome Atlas showed that most endometrioid tumors have few TP53 mutations or copy number changes but frequent mutations in KRAS, ARID1A, CTNNB1, PIK3CA, and PTEN and novel mutations in the ARID5B gene. Uterine serous tumors and high-grade endometrioid tumors had extensive somatic changes to chromosome copy number and frequent TP53 mutations, and a subset of endometrioid tumors had ultra-mutated POLE, encoding the catalytic subunit of DNA polymerase epsilon required for nuclear DNA replication and repair (14). Since the publication of the 2023 FIGO staging for endometrial cancer, molecular classification has been encouraged in all endometrial cancers. Molecular EC classification includes testing for pathogenic POLE mutation (POLEmut), mismatch repair deficient (MMRd) molecular subtypes, the non-specific molecular profile (NSMP) group, and TP53 abnormal (p53abn) mutations (15, 16).
The role of mitochondria (and mtDNA homoplasmy and heteroplasmy) in cancerMitochondria have several essential functions, such as steroid hormone biosynthesis, signaling, apoptosis, cell cycle control, production of energy, and so on (17, 18). Cells synthesize most of their ATP using the mitochondrial oxidative phosphorylation (OXPHOS) machinery, and this machinery requires 13 mtDNA-encoded proteins to function. Thus, proper mtDNA maintenance is essential to meet the basic energy demands within our cells. The multicopy mtDNA genome is replicated and repaired by the mtDNA polymerase gamma (Polγ) in concert with additional replisome factors, for example, Twinkle mtDNA helicase, topoisomerases, mitochondrial single-stranded DNA-binding protein, and others (19).
The polyploid human mtDNA genome is a covalently closed circular double-stranded 16,569-bp molecule that harbors the 13 OXPHOS genes mentioned above in addition to 2 genes encoding rRNAs, 22 tRNA genes and 8 genes that code for the mitochondrial-derived peptide signaling molecules (MDPs) (20, 21), Figure 1. The 24 RNA genes are required to translate the 13 mtDNA-encoded OXPHOS polypeptides. The MDPs are bioactive peptides with various physiological functions. For example, MOTS-c (Mitochondrial Open reading frame of The 12S rRNA-c) has been demonstrated to prevent diet-induced obesity and insulin resistance (23). MOTS-c is detectable in skeletal muscle and circulation and thus is described as a mitokine/mitochondrial hormone (24). MOTS-c translation occurs in the cytoplasm using the standard genetic code suggesting its RNA is exported from the mitochondria, while both mitochondrial and cytoplasmic expressed humanin have been suggested to be biologically active (20, 23).
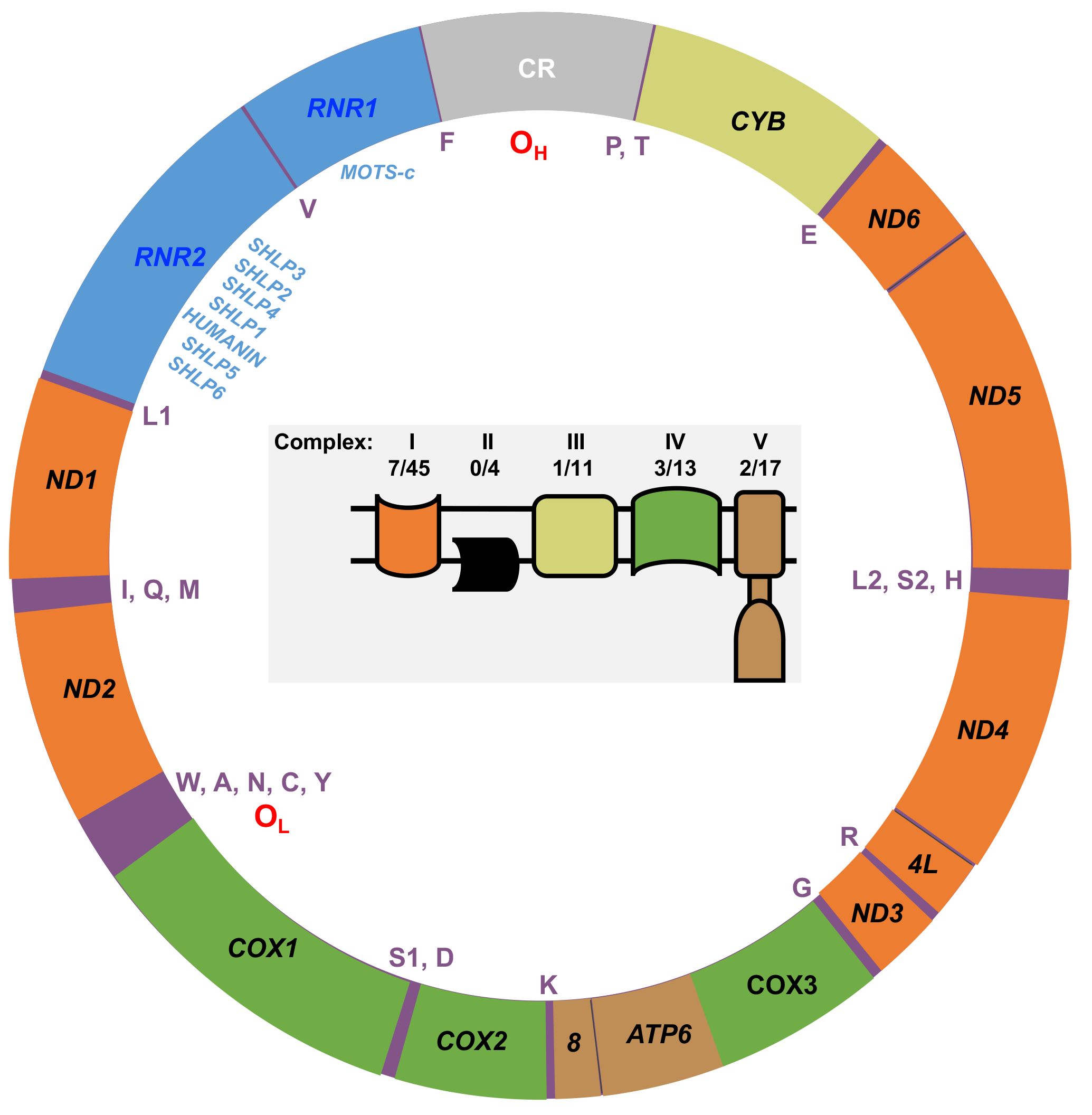
Figure 1 Map of the human mtDNA genome. The 13 genes encoding polypeptides of the mitochondrial OXPHOS machinery are labeled with black text on the map. Orange, OXPHOS complex I genes (NADH dehydrogenase, ND1, ND2, ND3, ND4L, ND4, ND5, and ND6); yellow, OXPHOS complex III gene (cytochrome bc1 complex cytochrome b, CYB); green, OXPHOS complex IV genes (cytochrome c oxidase, COX1, COX2, and COX3); brown, OXPHOS complex V genes (ATP synthase, ATP6, and ATP8). The small 12S (RNR1) and large 16S (RNR2) ribosomal RNA genes are colored blue, and the mtDNA control region (CR) is colored gray. The locations of the heavy strand origin of replication (OH), the light strand origin of replication (OL), the single letter amino acid residue codes for the 22 tRNA genes, and the eight mitochondrial-derived peptide (MDP) genes are indicated inside the map. The MDP genes localize within the RNR1 and RNR2 genes: HUMANIN, MOTS-c (Mitochondrial Open reading frame of The 12S rRNA-c), and the Small Humanin-Like Peptides 1 to 6 (SHLP1 – 6). Inside the mtDNA map is a cartoon of the mitochondrial inner membrane OXPHOS machinery. The OXPHOS complexes are color-coded according to their mtDNA gene colors on the map. Apart from complex II, the other OXPHOS complexes are encoded by both mtDNA and nDNA, and the number of mtDNA-encoded subunits out of the total number of complex subunits is indicated as previously reported (22).
Many cancers display aerobic glycolysis, also known as the Warburg effect (25). However, our current understanding is that tumor cells use both mitochondrial metabolism and increased glucose uptake and fermentation of glucose to lactate to synthesize ATP, macromolecules, and NADPH needed for rapidly proliferating cells (26, 27). In cancer cells, mitochondria provide the necessary building blocks for biosynthesis of amino acids, lipids, nucleotides, and glucose. During oncogenic activation, metabolism is rewired to generate ATP, and the needed citric acid cycle (CAC) intermediates utilized for macromolecule synthesis, e.g., citrate is exported from the mitochondria to the cytoplasm to generate the building blocks required for the biosynthesis of fatty acids and cholesterol (27–29).
Metabolic characteristics of tumors arise from cell-intrinsic factors like metabolic phenotype of the cell of origin and transforming genetic lesions and environmental factors such as the nutrients available in the tissue microenvironments. Regarding mitochondrial rewiring in cancer, two anaplerotic mechanisms have been observed. Anaplerosis replenishes intermediates removed from the CAC that were used to supply other biosynthetic pathways. In xenografts of colorectal samples, glutamine catabolism generates α-ketoglutarate (α-KG), Figure 2. In contrast, pancreatic and lung tumors favor pyruvate carboxylation via pyruvate carboxylase (PC) to replenish mitochondrial oxaloacetate (29). In pancreatic cells, PC and malic enzyme (ME1) are predicted to participate in a pyruvate cycle whereby cytosolic ME1 generates NADPH and pyruvate from malate. Next, the cytosolic pyruvate can enter the mitochondria and be converted to oxaloacetate via mitochondrial PC (30).
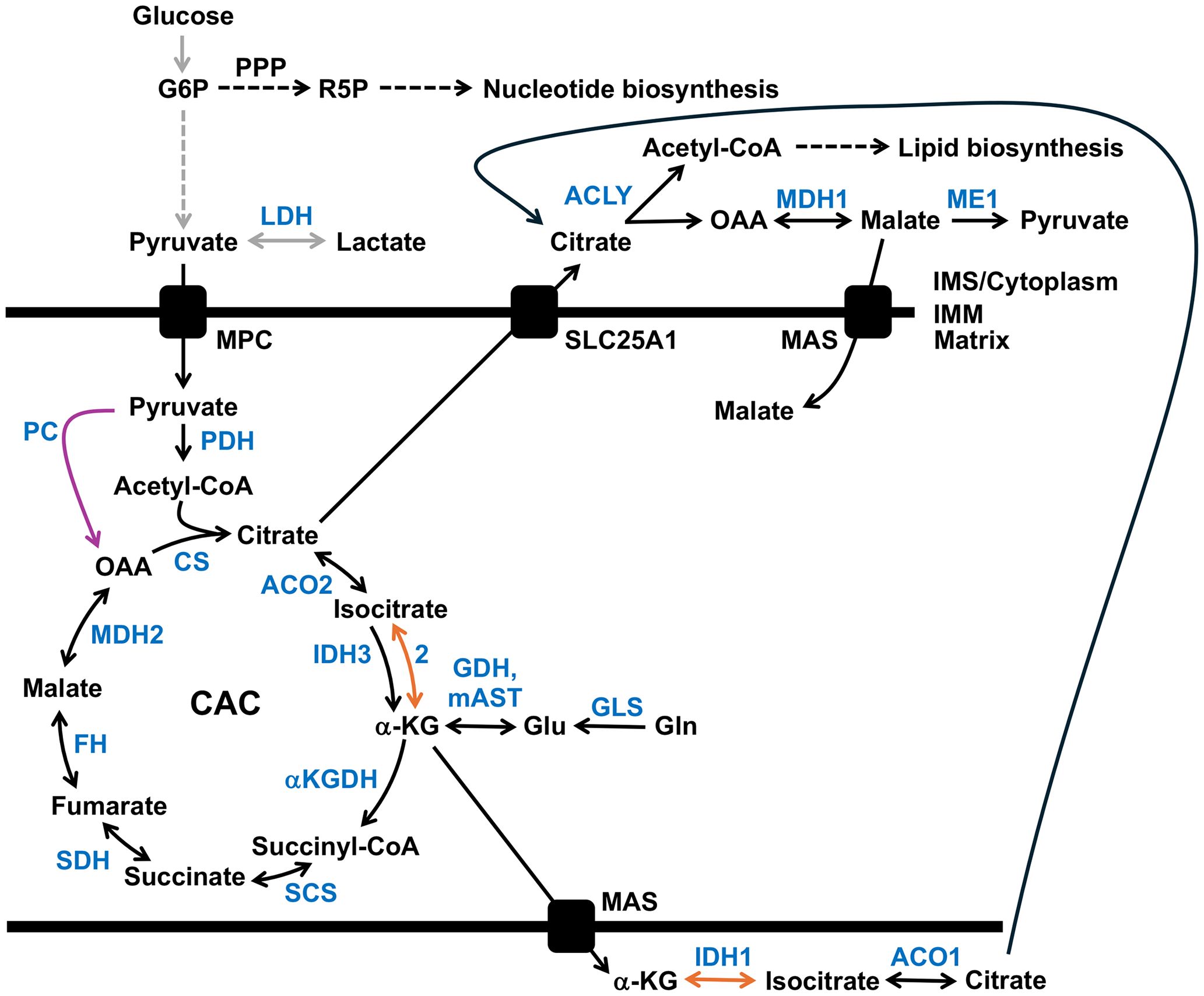
Figure 2 Metabolic adaptations in cancer. Gray arrows indicate the upregulation of glycolysis and enhanced lactate production, while the pyruvate carboxylase (PC) reaction is shown with a purple arrow. Reductive carboxylation of glutamine (Gln)-derived α-ketoglutarate (α-KG) to isocitrate is highlighted by orange arrows. Enzymes are highlighted in blue. See the text for details. ACLY, ATP citrate lyase; ACO1, cytosolic aconitase; ACO2, mitochondrial aconitase 2; αKGDH, α-ketoglutarate dehydrogenase complex; CAC, citric acid cycle; CS, citrate synthase; FH, fumarate hydratase (fumarase); G6P, glucose 6-phosphate; GDH, glutamate dehydrogenase; GLS, glutaminase; Glu, glutamate; IDH1, cytosolic isocitrate dehydrogenase; IDH2 and 3, mitochondrial isocitrate dehydrogenase 2 and 3; IMM, inner mitochondrial membrane; IMS, intermembrane space; LDH, lactate dehydrogenase; MAS, malate-aspartate (Asp) shuttle; mAST, mitochondrial aspartate aminotransferase; MDH1, cytosolic malate dehydrogenase 1 isozyme; MDH2, mitochondrial malate dehydrogenase; ME1, malic enzyme 1; MPC, mitochondrial pyruvate carrier; OAA, oxaloacetate; PDH, pyruvate dehydrogenase complex; PPP, the pentose phosphate pathway; R5P, ribose 5-phosphate; SCS, succinyl-CoA synthetase; SDH, succinate dehydrogenase complex; and SLC25A1, the dicarboxylate antiporter solute carrier family 25.
Although most cancer cells harbor functional mitochondria, tumors with mutations impairing and altering mitochondrial metabolism have been identified. In cancer, the mutation of genes encoding mitochondrial proteins, including isocitrate dehydrogenase 1 and 2, succinate dehydrogenase, and fumarase, have been documented (9). In patient-derived UOK262 renal carcinoma cells with mutations in the fumarase/fumarate hydratase (FH) gene or cells with a mutation in an OXPHOS complex I subunit, a reductive, glutamine-dependent pathway is the primary metabolic pathway. Similar to UOK262 (FH defective), 143B cells engineered to contain a loss-of-function mtDNA mutation in the complex III CYB gene had higher levels of citrate, malate, and fatty acids produced by reductive carboxylation of glutamine-derived α-ketoglutarate. Furthermore, using RNA interference, the authors showed that silencing the IDH1 (encoding the cytoplasmic isoform) or IDH2 (mitochondrial isoform) genes but not IDH3 (mitochondrial isoform) reduced the amount of glutamine-derived citrate implicating the IDH1 and IDH2 enzymes in the reverse reductive carboxylation reaction (31).
Could cancer cell heteroplasmic mutations in mtDNA similarly rewire metabolism? A recent study suggests yes and showed that murine cellular models of cancer-derived ND5 mtDNA heteroplasmic mutations regulate cancer metabolism and tumor biology, with redox imbalance contributing to a Warburg-like metabolic shift to glycolysis. Cell lines were engineered to separately contain the G11944A (human G12539A/W68Term, ovarian serous cystadenocarcinoma) and G12436A (human G13031A/p.W232Term, kidney renal clear cell carcinoma) mtDNA heteroplasmic ND5 termination mutations. Heteroplasmy-dependent increases in glucose-derived lactate and glutamine-derived cytoplasmic malate suggested the flow of reducing equivalents into the mitochondrion through the malate-aspartate shuttle (MAS) is impacted by changes in cellular redox. The increased abundance of glucose-derived lactate in heteroplasmic mutants was eliminated using the cytoLbNOX system to increase the NAD+/NADH ratio (32). The cytoLbNOX is a water-forming NADH oxidase genetic tool derived from Lactobacillus brevis that induces a cytosolic-specific increase of the NAD+ to NADH ratio in human cells. The human codon-optimized oxidase is specific for NADH rather than NADPH and catalyzes the reaction, 2NADH + 2H+ + O2 ➔ 2NAD+ + 2H2O (33).
Pyrimidine nucleotides play an essential role in metabolism, serving as RNA and DNA precursors, and uridine nucleotides act through receptors to regulate physiological processes (34). The DHODH gene is an essential gene that encodes the mitochondrial dihydroorotate dehydrogenase, an enzyme required for de novo pyrimidine base biosynthesis. The DHODH enzyme requires the functional activity of OXPHOS complex III (35, 36). The subunits of complex III are encoded by the CYB mtDNA gene and ten additional nuclear genes (37). As rapidly proliferating cancer cells have enhanced DNA replication and gene expression, glycolysis is likely increased to quickly generate ATP and intermediates needed by the pentose phosphate pathway to biosynthesize nucleotides. Augmented production of glycolytic ATP is predicted to provide an advantage to cancer cells as the rate of glycolytic glucose metabolism is 10-100 times faster than the complete oxidation of glucose through the mitochondria (26).
Furthermore, cancer cells display insensitivity to antigrowth signals, decreased autophagy, and impaired programmed cell death, i.e., apoptosis (28). In this model of rewired mitochondrial metabolism in cancer, mitochondria provide the essential TCA cycle intermediates needed by rapidly growing cells (38). Evidence for mitochondrial function in EC being critical for growth and proliferation comes from the observation that Type I (estrogen-dependent) patient samples demonstrate increased mitochondrial biogenesis compared to matched hyperplasia samples (39).
A cell can contain several thousand copies of circular mtDNA distributed within hundreds of individual mitochondria or throughout an elaborate mitochondrial reticular network (40). Human cancer cells harbor clonal synonymous (silent) and non-synonymous (non-silent, alters an encoded amino acid residue) mtDNA variants, so-called homoplasmic substitutions or homoplasmy. Additionally, cancer cells contain mixtures of wild-type (WT) and mutant mtDNA genotypes, known as mtDNA heteroplasmy (41–43). MtDNA mutation is a common feature of various types of cancer, and most solid tumors contain at least one mtDNA substitution (44). Mutations in mtDNA protein-coding genes can be silent or non-silent. Clinically critical variations in mtDNA can be categorized into three major classes: mtDNA germline mutations, somatic mutations, and ancient adaptive polymorphisms. Germline mutations are maternally inherited variants in the female germline; somatic mutations occur in oocytes, during embryogenesis, in somatic tissues, or in cancer tumors; mtDNA ancient adaptive polymorphisms result from selection during human migration (43).
mtDNA maintenance is essential to vertebrate lifeAll multi-cellular organisms require mitochondria for bioenergetics and biosynthesis of precursors for macromolecules (45). In some organisms, such as humans, specific short-lived cells like red blood cells, which live for ~120 days, lack mitochondria (and nuclei), presumably to reduce their size and prevent them from using the oxygen they carry (46). The maintenance of mtDNA is essential to vertebrate life. Knockout (KO) of the mouse POLG gene encoding the catalytic subunit of Polγ revealed embryonic lethality at E7.5–8.5 with subsequent depletion of mtDNA (47). Comparatively, several studies have illustrated the essential role of the Polγ processivity subunit (or accessory subunit) p55 in mtDNA replication: (i) two separate null mutations in the Drosophila melanogaster POLG2 gene lead to lethality in the early pupal stage of fly development (48), (ii) homozygous POLG2 KO mice are embryonic lethal at E8–8.5 (49) and (iii) in a porcine oocyte knockdown model, oocyte maturation requires POLG2 (50). Mouse RNaseH1-/- embryos are null at E8.5 and have decreased mtDNA content, leading to apoptotic cell death (51). A mouse model of Twinkle mtDNA helicase deficiency has been generated by transgenic expression of a Twinkle cDNA with an autosomal dominant mutation found in patients (52, 53). At one year of age, these mice developed progressive respiratory chain deficiency in cerebellar Purkinje cells, hippocampal neurons, and skeletal muscle. The affected cells accumulated multiple mtDNA deletions. These ‘Deletor’ mice recapitulate many of the symptoms associated with the POLG-related disease progressive external ophthalmoplegia and represent a valuable research model.
Our group recently generated another useful cell line research model of POLG-related mitochondrial disease. The human SJCRH30 myoblast cell line model harbors the most common autosomal dominant POLG mutation, c.2864A>G/p.Y955C, and displays bioenergetic deficits, decreased expression of OXPHOS complex I subunits, and impaired mtDNA maintenance (54). Furthermore, cancer cells lacking mtDNA fail to form tumors unless they reconstitute OXPHOS using mitochondria acquired from the host stroma (55). Therefore, mtDNA is essential to vertebrate biology and tumorigenesis.
mtDNA heteroplasmic mutations in cancerThe Cancer Mitochondria Atlas (TCMA) surveyed 2536 high-quality matched cancer and control sample pairs from the Pan-Cancer Analysis of Whole Genomes Consortium covering 38 specific cancer types and identified 7611 somatic mtDNA substitutions and 930 small indels. Of the 7611 variants identified in the whole-genome sequencing (WGS) data, >85% were heteroplasmic. Additionally, in contrast with nuclear DNA (nDNA) mutations where cancer type-specific signatures are seen, mtDNA mutations are similar across different tumor types, and most of the mutations display strand bias with predominantly G > A and T > C substitutions on the L-strand (17).
A recent study utilized a sensitive whole-exome sequencing (WES) method focusing on tRNA, rRNA, and protein-coding genes. The study determined that predicted pathogenic mtDNA mutations arise in tumors at a rate similar to mutations in the standard cancer driver genes. However, it should be kept in mind that mtDNA mutations located in regions without adequate sequencing coverage are not identifiable with repurposed WES data, and this data can be biased towards variants with elevated heteroplasmy. In this study, 3,264 matched tumor and normal samples had sufficient coverage to call mutations in at least 90% of the mtDNA genome, and of these, fifty-seven percent harbored at least one mtDNA variant. Interestingly, predicted pathogenic mtDNA mutations were shown to be associated with increases in the survival of colorectal cancer patients. The CYB cytochrome b gene showed increased rates of missense mutations, complex I genes were shown to accumulate loss-of-function mutations at homopolymeric runs at an increased rate, and complex V genes were depleted of non-synonymous mutations, implying negative selection against ATP synthase gene mutations. Also, the transcriptional analysis from this study showed truncating mtDNA mutations promote decreased expression of innate immunity genes and increased expression of OXPHOS genes (56).
To identify somatic mutations in cancer, we assume a particular mutation present in the cancer tissue will be absent in the normal tissue of the same individual (17, 57). However, the effect of most of these somatic mtDNA mutations and their exact role in carcinogenesis remains unclear. MtDNA mutations have been found to hinder energy production, cause metabolic disorders, and alter the production of ROS (58). The rate of mutation in mtDNA genes is estimated to be 100- to 1000-fold higher than nDNA genes, which makes mtDNA a good candidate for involvement in cancer progression (59, 60). It is reasonable to conceive that cancer mtDNA mutations can be either driver mutations, providing a selective growth advantage to a cell, or passenger mutations, not providing any advantage. Considering the increase in cancer cell energy demand compared to normal cells, functionally relevant driver mtDNA mutations in cancer seem plausible (61). Furthermore, mtDNA de novo mutations are hypothesized to function as either strong ‘inducers’ of carcinogenesis or mild ‘adaptors’ that permit a cancer cell to adapt to different environments (43). As the mtDNA and nDNA genomes encode subunits of the OXPHOS machinery, mutations in either genome could alter ROS production or the cell’s redox status, contributing to tumor growth (62). Changes in mitochondrial genes encoded on either genome could alter mitochondrial metabolites, further altering gene expression and contributing to tumor growth (43).
Further evidence demonstrating the essentiality of vertebrate mtDNA comes from the hundreds of documented pathogenic mtDNA mutations linked with multisystem degenerative disorders (60). Therefore, in cancer, it has been hypothesized that neutral missense mutations drift toward homoplasmy while deleterious or pathogenic mutations are under negative selection to remove harmful mutations (63). Suppose cancer cell mtDNA mutations are predicted to be detrimental if homoplasmic; why are they not removed by purifying (negative) selection, and is there some selective advantage to a cancer cell maintaining a predicted pathogenic mtDNA mutation in heteroplasmy? In cancer cells, we expect that mitochondria are under selective pressure to keep the mtDNA-encoded OXPHOS subunits. Therefore, if a mtDNA gene is mutated to alter the function of its encoded protein detrimentally, then the organelle can compensate by maintaining a mixed population of WT and mutant mtDNA molecules (57). We hypothesize that EC cells benefit from carcinogenesis-inducing or environment-adapting heteroplasmic somatic mtDNA mutations (predicted pathogenic) that favor EC metabolism, hereafter inducing/adapting mutations. On the other hand, we expect more mild (less damaging or pathogenic) environment-adapting mutations could drift towards mtDNA homoplasmy.
EC-specific mutations occur across the mtDNA genomeNext, to understand EC mtDNA mutations that occur across the entire mtDNA genome, we review mtDNA data obtained from Mseek and WGS studies. The tumor-specific mtDNA mutations, heteroplasmy levels, and the mutations’ predicted impacts are discussed below. Previously, we used the Mseek mtDNA-specific next-generation sequencing (NGS) approach to investigate the entire mtDNA from 3 matched sample sets (tumor and peri-normal) and identified nine single nucleotide somatic EC mtDNA variants. Mseek exploits mtDNA circular topology and treatment of DNA samples with exonuclease V to reduce the amount of nuclear genomic reads. The coverage of our Mseek data was >400x (57). One hundred and ninety-one somatic EC mtDNA variants from 102 sample sets were obtained from the NGS study by Grandhi et al. that utilized data accessed from The Cancer Genome Atlas (TCGA) August 2015. Tumor and matched normal sample sets were compared to determine an appropriate uniform read depth to detect somatic mutations, >50x (64). One hundred forty-five variants from 40 samples were obtained from the ultra-high depth Yuan et al. NGS study, with >5,000x average coverage for uterus adenocarcinoma samples. The group extracted mtDNA sequence data from the whole-genome alignment files of cancer samples and their matched normal tissue samples obtained from The Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium. The PCAWG aggregated WGS data from the International Cancer Genome Consortium (ICGC) and TCGA projects (17).
The Grandhi et al. and Yuan et al. studies both mined data from TCGA, but at different points in time and using different bioinformatics methods to call mtDNA variants and heteroplasmy. Therefore, we carefully scrutinized the results to remove likely duplicated samples with identical mutations and highly similar levels of heteroplasmy. On average, the duplicates had a difference of 2.1% heteroplasmy. Based on the duplicate analysis, we removed 34 samples with 66 mutations from the set obtained from the Grandhi et al. study, and the matched mutations in the Yuan et al. study were kept in the analysis. The result is 279 EC tumor-specific single nucleotide variants from 111 samples (an average of 2.5 variants per sample), Supplementary Table 1 (17, 57, 64). Seven recurrent mutations were identified within this set of variants, three from the Grandhi et al. and four from the Yuan et al. studies, Figure 3.
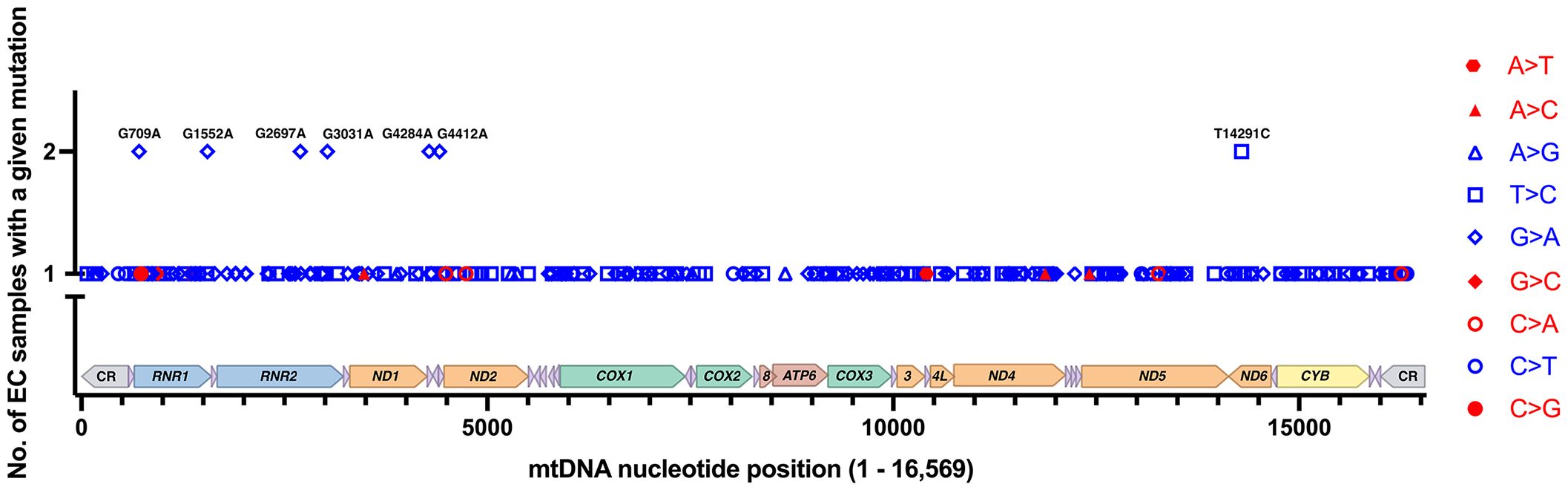
Figure 3 EC-specific mutations and their positions on a linear map of the mtDNA genome. Of the 279 mutations from 111 samples, 272 are unique, and 7 reoccurred in separate samples. Transitions are shown in blue, and transversions are in red. Each type of mutation is represented with a different symbol. The mutations shown on the graph are changes from the rCRS NC_012920.1. The mtDNA genes are colored as in Figure 1.
According to the strand displacement model of mtDNA replication, replisomes containing Polγ synthesize both the nascent heavy (H) and light (L) strands continuously without the formation of Okazaki-fragment-like replication products (65). The two mtDNA strands are named H and L based on the ability to separate them on denaturing cesium chloride gradients. The H-strand is richer in G+T content, making it heavier on density centrifugation (66, 67). The origin of H-strand DNA replication (OH) is located in the non-coding control region (CR), and the origin of L-strand replication (OL) is located ~11,000 base pairs downstream of OH, Figure 1 (65). The CR harbors a three-stranded DNA zone known as the displacement loop or D-loop (68, 69). Another difference from nDNA replication is that mtDNA molecules are replicated independently of the cell cycle (70). During mtDNA replication, relatively long single-stranded stretches of the polyploid H-strand templates are formed, which can be argued to be sensitive targets of exogenous and endogenous damage and need protection during the replication process by the mitochondrial single-stranded DNA binding protein, mtSSB (54).
The EC-specific mtDNA variants represent changes from the revised Cambridge Reference Sequence (rCRS). Out of the 279 EC tumor-specific mtDNA mutations, transitions were 24.4-fold higher than transversions (268 transitions and 11 transversions), Figure 3. The high transition: transversion ratio (ts/tv) has been previously noted in human mtDNA, with transitions being 15-fold higher than transversions (71); however, in EC tumors, the ratio is even higher. EC-specific mutations occurred across the mtDNA genome in both non-coding regions (CR, NC5, and OL) and coding genes except for ten tRNA genes (TRNF, Q, W, A, N, Y, H, L2, E, and T), which did not harbor any mutations. Of the total mutations, 65.9% occur in protein-coding genes, 7.5% in tRNA genes, 20.4% in rRNA genes, and 6.1% in the non-coding regions. The mutations in protein-coding genes were both synonymous and non-synonymous. About one-quarter of the 57 mutations in the rRNA genes localize to the MDP open reading frames. These consisted of a synonymous MOTS-c mutation, three silent mutations in RNR2 MDP genes (SHLP5, SHLP6), and ten non-synonymous mutations in RNR2 MDP genes (SHLP3, SHLP1, Humanin, SHLP5, and SHLP6).
EC mtDNA mutations reoccurSeven EC-specific mtDNA transition mutations reoccurred in more than one sample, and these comprised 14 of the 279 mutations, G709A (RNR1), G1552A (RNR1), G2697A/R22K (Humanin/RNR2), G3031A/K14K (SHLP6/RNR2), G4284A (TRNI), G4412A (TRNM), and T14291C/E128G (ND6), Figure 3. Eleven of the 14 recurrent mutations fell between 10 and 90% heteroplasmy except for one of each of G2697A (90.02% heteroplasmy), G3031A (96.74%), and T14291C (3.95%), Supplementary Table 1. Unfortunately, there is currently no sufficient model to predict a mutation’s pathogenicity when it occurs in the mtDNA rRNA genes (RNR1, RNR2), the short open reading frame MDP signaling molecules (humanin, MOTS-c, and SHLP1 – 6), or the non-coding regions (CR, NC5, and OL). The MITOMASTER mtDNA sequence analysis tool (72) revealed that apart from the frequent G709A variant (present in the three L/African, M/Asian, and N/Eurasian human mtDNA Lineages in Mitomap at 11.32%, 9.87%, and 14.31% respectively), the remaining recurrent variants were present at 0% or equal to or less than 0.007% in the GeneBank, gnomAD (73), and HelixMTdb (74) reference population databases. Furthermore, a significant amount of the total 279 variants were 0%, or there was “No Record” in the GeneBank, gnomAD, and HelixMTdb databases, 56% (156 variants), 63% (176 variants), and 58% (161 variants), respectively. However, care needs to be taken when assessing variants as a rarity in the population should not be considered the only line of evidence for a mutation’s pathogenicity as many unrepresented variations are benign and databases are far from saturation (73).
Using the Mitochondrial tRNA Informatics Predictor (MitoTIP) in silico tool (75), the TRNI G4284A mutation is predicted to be possibly benign but is associated with spastic paraparesis (76). The TRNM G4412A mutation is expected to be likely-pathogenic and is associated with seizures, myopathy, and retinopathy (77); therefore, when present in heteroplasmy at 27.63 or 66.85%, the G4412A tRNA mutation is predicted to be an inducing/adapting mutation that alters EC cellular metabolism. The silent SHLP6 G3031A/K14K mutation is expected to be non-harmful, but as mentioned previously, the effect of the mutation on RNR2 cannot currently be predicted.
Non-synonymous mutations were categorized into predicted inducing/adapting mutations (predicted pathogenic) and non-pathogenic groups. Utilizing the MitImpact database collection of genomic, clinical, and functional annotations for non-synonymous mutations in mtDNA protein-coding genes (78), we scored a mutation as a predicted inducing/adapting mutation that is potentially able to favor EC cell metabolism if at least two out of three pathogenicity predictor scores were deleterious [CAROL, Combined Annotation scoRing tool (79)], likely-pathogenic/pathogenic [APOGEE2, pAthogenicity Prediction thrOugh loGistic modEl trEe (80)], and medium/high-impact [Mutation Accessor (81)]. Although the T14291C/E128G (ND6) variant is absent from the GeneBank, gnomAD, and HelixMTdb databases, the in-silico pathogenicity scores predict it is likely non-pathogenic. Of the variants shared among two patients described above, four occurred in the rRNA genes. Also, two RNR2 gene mutations encode mutations in MDP genes, one is non-synonymous (Humanin G2697A/R22K), and the other is silent (SHLP6 G3031A/K14K).
Thus, out of the 279 EC mtDNA mutations identified in 111 patients, 265 occurred in single samples (do not reoccur), and 272 unique mutations were identified in the cohort. Of the 265 non-reoccurring variants, 49 localize in rRNA genes and 17 in non-coding regions. For the remaining non-reoccurring variants, 83 are predicted to be inducing/adapting mutations (termination mutations, non-synonymous mutations, and pathogenic tRNA changes), 61 are non-synonymous non-pathogenic, 8 are tRNA non-pathogenic mutations, and 47 are silent substitutions. Next, we consider the 279 mutations in aggregate to gain insight into the current collection of tumor-specific mtDNA mutations identified in the EC cohort.
mtDNA transitions are the most abundant mutations in EC tumorsAs mentioned above, EC tumor-specific transition mutations were 24.4-fold higher than transversions. The increased level of EC mtDNA transitions agrees with a study of 1907 mtDNA mutations from 31 different cancer types that found transitions represent most of the substitutions (61). Furthermore, Ju et al. found strand bias concerning these transition mutations, with 76.8% of the T>C mutations on the L-strand (TL:AH to CL:GH) and 84.1% of the C>T variants on the H-strand (GL:CH to AL:TH). The strand bias is present in the EC samples, with 91.4% of the T>C mutations on the L-strand and 86.8% of the C>T variants on the H-strand. Transition mutations were the most abundant changes in the EC tumor mtDNAs when normalized to all other mutations, with 47.3% GL:CH to AL:TH and 38.0% TL:AH to CL:GH, Figure 4. These transitions likely arose from 1. H-strand C to U deamination events and 2. erroneous incorporation of a nascent H-strand G across from a L-strand template T by the replicative Polγ followed by subsequent rounds of mtDNA replication resulting in CH>TH and AH>GH, respectively, and as previously suggested (61, 82, 83). We propose the single-stranded nature of replicating mtDNA drives the accumulation of CH>TH mutations in cancer. This is because C>T transitions resulting from spontaneous deamination of C to U have been shown to occur at orders of magnitude more quickly in ssDNA than dsDNA (84), and because replication intermediates harbor significant stretches of H-strand ss-mtDNA. Furthermore, because Polγ is prone to inserting G across from a template T, and as estrogen stimulates the biogenesis of mitochondria (9), the high levels of EC mtDNA AH>GH mutations likely represent signatures of increased mtDNA replication in cancer.
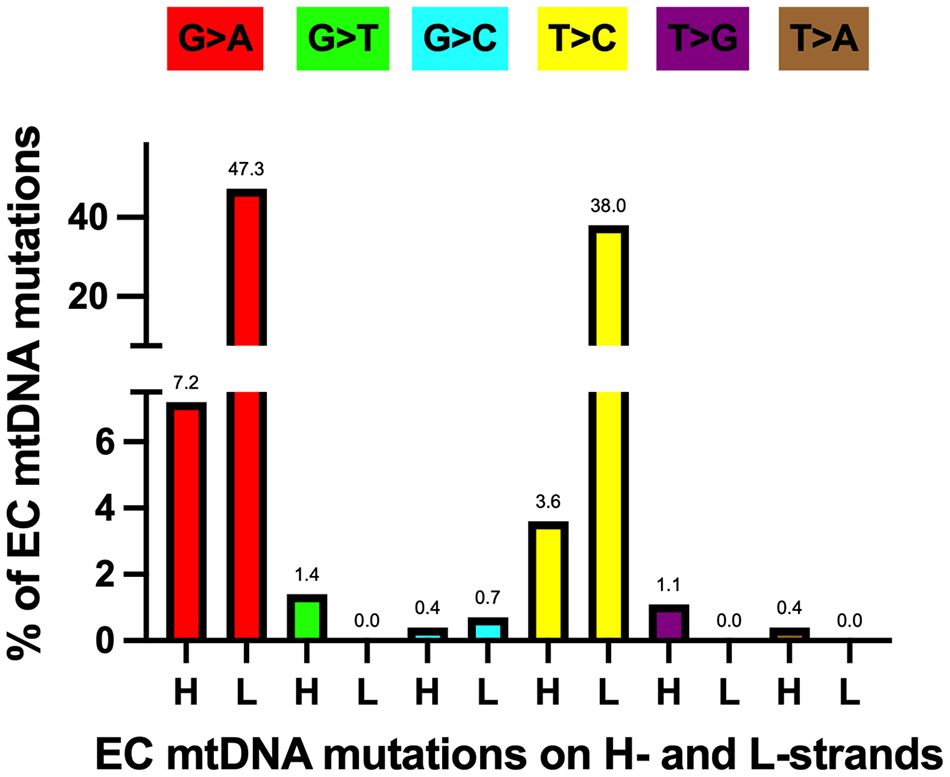
Figure 4 Most EC-specific G>A and T>C transition mutations occur on the mtDNA light (L) strand. H, heavy strand. Each mutation is shown as a percentage of the total mutations. Different colored bars represent each substitution type; the mutation type is shown above each bar. Based on data from references (17, 57, 64), see Supplementary Table 1.
The subsequent most abundant mutations were 7.2% GH:CL to AH:TL and 3.6% TH:AL to CH:GL and these could have arisen at short single-stranded regions of the replication fork by similar means as described above but on the opposite strand. EC tumor transversion mutations were low, with GH:CL to TH:AL (1.4%) and TH:AL to GH:CL (1.1%) being the most abundant. Due to the proximity of the mtDNA nucleoid to the OXPHOS machinery, we hypothesize that the GH>TH mutations result from ROS-induced deoxyguanosine base damage in the form of 7,8-dihydro-8-oxo-2’-deoxyguanosine, 8-oxo-dG (85). In this scenario, Polγ would mis-incorporate an A across from the H-strand template containing the 8-oxo-dG base, resulting in a G>T transversion following mtDNA replication. Finally, TL:AH to AL:TH, TL:AH to GL:CH, and GL:CH to TL:AH transversions were not detected in the EC tumors.
Half of the tumor samples harbor mtDNA mutations predicted to favor EC metabolismWe predict that out of 279 mtDNA substitutions, 63 are non-synonymous inducing/adapting mutations (22.6%), and another 22.6% are non-synonymous and non-pathogenic, Figure 5. Eleven substitutions occurring in tRNA genes are predicted to be inducing/adapting mutations (3.9%), and 10 are likely non-pathogenic (3.6%). There are 11 mutations in the protein-coding genes that formed termination or nonsense codons (3.9%), and of these, 10 are GL:CH>AL:TH. We expect that the mtDNA termination mutations are inducing/adapting mutations that can favor EC metabolism as they are predicted to alter their respective protein’s function. Utilizing the MitImpact database, we determined the impact of changing the amino acid residues removed because of the various termination codons. In all these cases, changing the amino acid residues at the termination positions is predicted to alter metabolism, as the other two MitImpact SNVs considered at these positions were predicted to be impactful, deleterious, and pathogenic with at least two out of three pathogenicity predictor scores as described above. Moreover, changing a sense codon to a stop codon can lead to the loss of many amino acids and the complete protein will not be formed. In our analysis, these nonsense mutations truncated 11.6% to 99% of the proteins, with an average of 55% being truncated. The lost amino acid residues are likely essential to the function of their respective protein. Six of the 11 unique nonsense mutations localize to complex I genes (one mutation in ND4 and five in ND5), which agrees with the previous observation that truncation mutations preferentially arise in complex I genes relative to other genes (56).
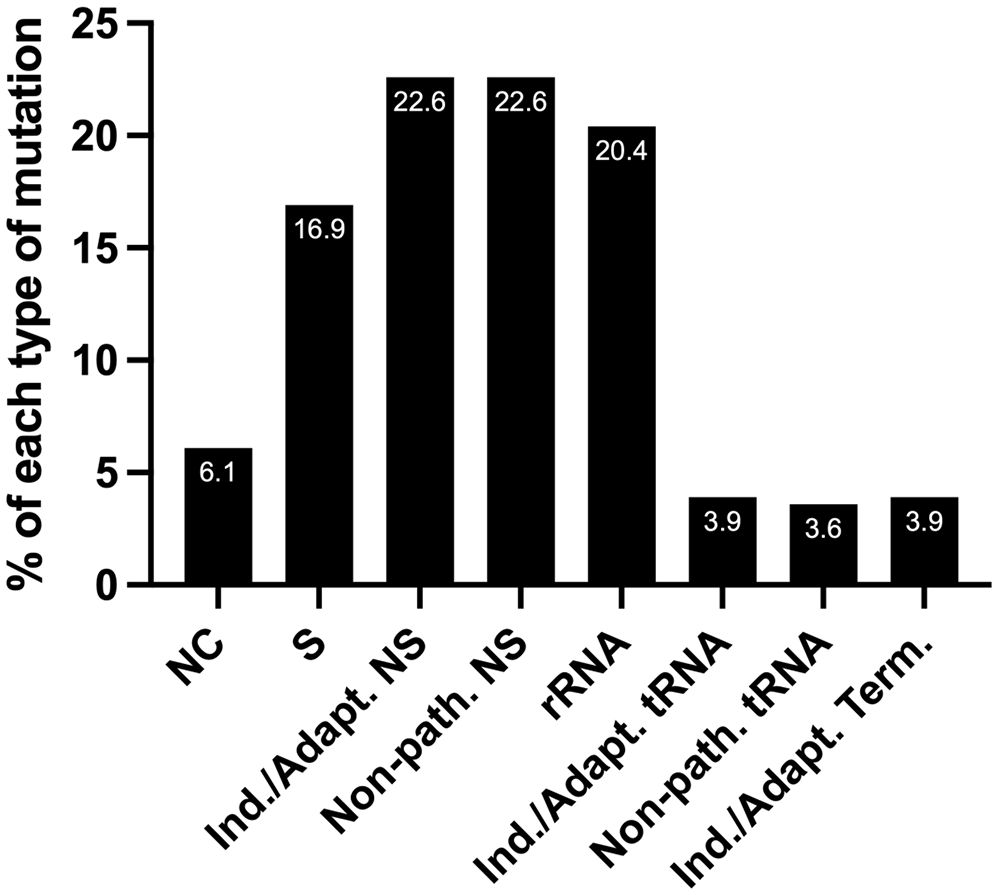
Figure 5 The percentage of each type of mutation from the set of 279 EC mtDNA somatic substitutions. NC, non-coding; S, synonymous (silent); NS, non-synonymous (non-silent). Nearly a third of the substitutions are predicted carcinogenesis-inducing or environment-adapting mutations that favor EC metabolism (Ind./Adapt.); Non-path., predicted non-pathogenic; Term., termination; see the text for details.
Defective complex I in cancer has been proposed to potentiate metastasis by enhancing ROS production, promoting tumor formation, and increasing resistance to cell death stimuli. Additionally, ND5 gene mutations are linked to different cancers like colon adenocarcinoma, acute myeloid leukemia, breast cancer, and myelodysplastic syndrome (a.k.a. MDS, preleukemia), and these changes are suggested to inhibit OXPHOS resulting in altered mitochondrial bioenergetics which could confer a selective growth advantage to cancer cells (86). In terms of the 111 EC tumors with 279 tumor-specific mutations, 107 mutations occur in complex I genes, and the majority occur in ND5 (11 in ND1, 14 in ND2, 9 in ND3, 24 in ND4, 3 in ND4L, 36 in ND5, and 10 in ND6). Of these variants, six result in termination mutations, 28 are silent, and 73 are non-silent. Thirty-three of the non-silent mutations are predicted to be inducing/adapting mutations, and 40 are expected to be non-pathogenic.
For 279 mutations, heteroplasmy ranged from 1 to 98.8%, averaging 49.2%. As we hypothesize that tumors could benefit from heteroplasmic mtDNA inducing/adapting mutations that favor EC metabolism, we looked at tumors that harbored at least one mtDNA mutation allele at 10% (WT 90%) to 90% (WT 10%) heteroplasmy (assuming slight to significant changes in EC tumor metabolism, respectively). We found that 79.3% of the 111 tumor samples contained at least one heteroplasmic mutation within this range, Supplementary Table 1.
Next, we wondered how many patients harbored predicted inducing/adapting mutations because they represented nearly a third of the total mutations (22.6 + 3.9 + 3.9%), Figure 5. Interestingly, half of the patient samples (56/111) harbored a predicted inducing/adapting mutation, which is significant considering it is currently not possible to predict the effects of a similar proportion of rRNA (20.4%) and non-coding (6.1%) mutations. The remaining 16.9% of the EC mtDNA substitutions are silent variants, Figure 5.
Of the 85 predicted inducing/adapting mutations, 46 have heteroplasmy levels between 10 and 90% (including 7 of 11 termination mutations and 9 of 11 tRNA mutations). Twenty-three non-synonymous predicted inducing/adapting mutations were <10% heteroplasmy, while 16 had >90% heteroplasmy (2 tRNA and 14 non-synonymous mutations in protein-coding genes). We expect the 16 with greater than 90% heteroplasmy could be mild environment-adapting mutations drifting towards homoplasmy. On the other hand, we speculate that the 23 non-synonymous predicted inducing/adapting mutations with less than 10% heteroplasmy may have been isolated from low-grade tumors or tumors surgically removed earlier during carcinogenesis. There were 3 predicted inducing/adapting termination mutations with less than 10% heteroplasmy, and 1 with greater than 90% heteroplasmy, and 2 tRNA inducing/adapting mutations with >90% heteroplasmy, Supplementary Table 1.
The 85 predicted inducing/adapting somatic mtDNA mutations exist among 56 tumor samples, suggesting there could be synergistic effects on metabolism when different mutations are in the same tumor. Approximately 59% of the tumor samples (33 of 56) harbored at least one predicted inducing/adapting mutation that falls between 10 and 90% heteroplasmy (12 tumors harbored predicted inducing/adapting mutations at <10% heteroplasmy and 11 had mutations with >90% heteroplasmy). Thirteen of the tumor samples harboring predicted inducing/adapting mtDNA mutations outside the 10 to 90% range also had mutations in either the rRNA genes or the non-coding regions. Therefore, 30% of the total 111 tumor samples harbor a predicted inducing/adapting somatic mtDNA mutation between 10 and 90% heteroplasmy, suggesting these variants could play an essential role in EC metabolism.
EC mtDNA insertions and deletions reoccur in different samplesTwenty-eight mtDNA indels (16 insertions and 12 deletions) in 25 EC samples were identified, Supplementary Table 2. Eighteen of the samples with indels also had mtDNA somatic mutations (i.e., 18 of 25 samples with indels were also part of the 111-sample cohort with 279 total somatic mutations). Heteroplasmy was not reported for 18 of 28 indels, but the remaining 10 had heteroplasmy ranging from 16 to 95%, with an average of 52%. Approximately 68% of the indels occurred in coding regions (19 mutations), and ~54% occurred specifically in complex I genes. 79% of the indels were single nucleotide insertions or deletions. Although the pathogenicity predictor tools do not predict the impact of indels, we hypothesize that frameshift mutations in the coding region of a mtDNA-encoded protein will be inducing/adapting mutations that alter the protein’s function and, by extension, the cell’s metabolism. The three nucleotide in-frame deletion TAGC12989T does not disrupt the reading frame but does delete the evolutionarily conserved A219 amino acid residue of ND5. Six indels occurred in non-coding regions (4 in the CR, 1 in NC5, 1 in NC7), one in RNR2, and two in tRNA genes, TRNS1 and TRNP.
Five EC mtDNA indels in complex I genes reoccurred in more than one sample, comprising 11 of 28 indels (39%). Of the five reoccurring indels, the ND4 A11866AC insertion is shared among three individuals, while the remaining variants are shared among two individuals, ND1 A3565AC, ND4 A10946AC, ND5 CA12417C, and ND5 C12417CA. Interestingly, more than half of the reoccurring mtDNA indels, A11866AC (m.11872insC), CA12417C (m.12425delA), and A3565AC (m.3571insC), have been reported to likely induce oncocytoma (43). Therefore, these mutations may also be inducing/adapting mutations that favor EC metabolism and could serve as biomarkers for EC detection. Of the 28 EC mtDNA indels identified in 25 samples, 17 do not reoccur, and 22 unique indels were identified in this cohort.
mtDNA copy number changes in ECHuman mtDNA occurs at a high copy number and varies between cell types, ranging from >150,000 to ~100,000 copies in mature oocytes, 1000s of copies per muscle fiber, and an average of 100 copies per sperm cell (87–89). There are interindividual variabilities in mtDNA copy numbers in human cells, which have been suggested to be linked to pathological features like obesity and cancer (90). Studies have shown that a high mtDNA copy number in whole-blood DNA extracts is linked to lung and breast cancer risk (91, 92). In contrast, low mtDNA copy number in whole-blood DNA extracts is associated with risks of renal cell carcinoma, soft tissue sarcoma, and esophageal adenocarcinoma (93–95). Another study showed that mtDNA from whole-blood DNA extracts is present at lower levels than in controls and is associated with an increased risk of endometrial cancer (90). Additionally, mtDNA content increased in hyperplastic and EC tissues compared to control tissue (96). In different studies with n ≥32 control samples and n ≥40 EC samples, 2- to 2.6-fold increases in mtDNA content were found in EC tumors compared to normal tissue (97, 98). In a study carried out by our group looking at three EC patients’ matched tumor and peri-normal tissues, we found that mtDNA copy number was increased relative to peri-normal tissue in one patient, decreased in another, and did not change in the third individual. In the same patients, mtDNA topological isomers (topoisomers) were studied, and catenated species were increased in the three EC tumor samples relative to the matched peri-normal specimens, suggesting enhanced mtDNA replication in the tumors (57). Interindividual variabilities in mtDNA could explain the lack of increased mtDNA copy number levels in particular EC samples in the various studies.
mtDNA haplogroups and ECSubstantial mtDNA sequence diversity exists between individuals and human populations. Evidence suggests that ancient mtDNA polymorphisms accumulated along maternal lineages as humans migrated out of Africa. If a mutation changed mitochondrial physiology to benefit individuals within that environment, then that variant became enriched in that location. Further mutations in descendant mtDNA genomes generated a group of related regional mtDNA variant genotypes known as haplogroups. Therefore, each continent and geographical region is associated with characteristic mtDNA haplogroups (60). Haplogroups A, B, C, D, and E are specific to Asian populations, and one study investigated the relationship between EC and mtDNA variation in subjects with a Han native background in southwestern China. Using Chi-square statistics, frequency differences of haplogroups were tested between an EC group (n = 49 cancer patients) and a control group (31 control individuals). The study found that EC patients clustered in haplogroup D with a significantly higher frequency than controls (χ2 = 5.685, P = 0.017), suggesting a possible association of haplogroup D to EC. Additionally, the mtDNA haplogroup D C5178A allele frequency between EC patients and control subjects was significant (χ2 = 7.143, P = 0.007), suggesting it could be linked to EC pathogenesis (99).
Haplogroups H, I, J, K, T, U, V, W, and X are distributed among the European populations. In a study conducted in Lublin, Poland, mtDNA sequences of 26 EC patients were compared with the general Polish population to test for an association with cancer susceptibility. The Haplogroup H C7028T polymorphism was strongly underrepresented (χ2 = 8.58, P = 0.003) in three EC patients relative to the general Polish population suggesting haplogroup H could be a cancer-protective group (100). Future haplogroup studies with significant numbers of EC patients and general population mtDNA sequence data are needed to understand better how regional populations and their haplogroups are at risk of developing EC.
Treatment of ECCommon treatments for cancer include surgery, radiation therapy (RT), and chemotherapy. Standard EC treatment involves surgery, i.e., the removal of the uterus, cervix, fallopian tubes, and ovaries, and selective pelvic and para-aortic lymphadenectomy. Women who are not candidates for surgery can be recommended RT. Additionally, progestin-containing intrauterine devices (IUDs) can be used as an alternative for young individuals wishing to preserve fertility, as these devices have been demonstrated to regress endometrioid EC (101–103). Following treatment, patients may undergo hormonal therapy, RT (external beam and/or vaginal brachytherapy), or chemotherapy, depending on their risk factors and stage of disease. Hormonal therapy has been primarily evaluated in low-grade endometrioid cancers. Agents include Megace (or megestrol, a progestin of the 17α-hydroxyprogesterone group) alternating with tamoxifen, progestational agents, and aromatase inhibitors. Good responses have been seen in patients with ER/PR-positive disease, low-grade disseminated disease, and pulmonary metastases (104–112). Chemotherapy treatment for metastatic EC can utilize single or multiple agents, including taxanes (e.g., paclitaxel), anthracyclines (e.g., doxorubicin), and platinum compounds (e.g., cisplatin) (11). These treatment options are highly individualized (and beyond the scope of this review).
First-line therapies have moved beyond paclitaxel and carboplatin (another platinum-containing compound) and now include immunotherapy agents in combination with chemotherapy. Humanized antibody pembrolizumab and monoclonal antibody dostarlimab are approved immune checkpoint inhibitors used in treating patients with recurrent or primary advanced EC that are either deficient (MMRd) or proficient (MMRp) in DNA mismatch repair. During nDNA synthesis, the incorporation of an incorrect nucleotide sometimes occurs, and cells use MMR protein machinery to repair these errors. Because MMRd EC tumors produce abnormal variant proteins, they tend to attract immune cells. Two recent large, randomized trials, NRG-GY018 (113) and RUBY (3) have shown a progression-free survival benefit by adding pembrolizumab (NRG-GY018) or dostarlimab (RUBY) to paclitaxel and carboplatin in patients with stage III or IVA endometrial carcinoma with measurable disease, or stage IVB or recurrent disease of any histologic subtype, (except for carcinosarcoma for pembrolizumab). Significantly, patients with either MMRd or MMRp benefited from these combinations, with the greatest benefit seen in patients with MMRd tumors.
Exploiting cancer cell mitochondria for cancer therapyMitochondria are increasingly considered targets for cancer therapy due to their essential role in programmed cell death (i.e., apoptosis), cellular metabolism, and cell signaling (114). Researchers have focused on designing compounds that function as mitochondrial-targeting ligands and carry anticancer agents to the organelle. Examples of mitochondrial-targeting ligands include triphenylphosphonium (TPP, a cationic molecule that penetrates through mitochondrial membranes and accumulates in mitochondria) attached to the anticancer DNA damaging agent chlorambucil (chlorambucil linked TPP) and cationic mitochondria-penetrating peptides (MPPs) attached to anticancer drugs (e.g., doxorubicin linked MPP) (28). Cancer therapies that target mitochondria can inhibit energy supply to kill cells. Also, compounds that interfere with the electron transport chain, a significant site of ROS production, can increase the risk of electron leakage and ROS production, which can then damage the mitochondria and lead to apoptosis.
Mitochondria also play an essential role in ferroptosis, an iron-dependent form of non-apoptotic cell death driven by lipid peroxidation due to iron accumulation. Like apoptosis, ferroptosis induces decreased mitochondrial membrane potential but, unlike apoptosis, does not require caspase activation. In a study using HepG2 and Hep3B liver cancer cell lines, the CDGSH iron sulfur domain 1 iron-containing outer mitochondrial membrane protein (CISD1, also named mitoNEET) was shown to inhibit ferroptosis by protecting agains
留言 (0)