Animals
A total of 81 wildtype C57BL/6J mice (male, 8 weeks old), provided by the Laboratory Animal Center of the Fourth Military Medical University [Xi’an, People’s Republic of China, license No. SCXK (Shan) 2019-001], were employed in the present study. All the animals could get free access to water and food and accommodated under a satisfactory environment [12-h light/dark cycle, controlled temperature (22 ± 1 ℃), and humidity (60 ± 5%)]. Most importantly, strict ethical guidelines for the pain investigation were needed, and the protocol (No. IACUC-20,211,101) was authorized by the local Ethics Committee on Animal Application for Research and Education of the Fourth Military Medical University.
This study can be divided into three parts. Firstly, we identified the location of Rab11a in the SDH. Then, chronic inflammatory pain model was constructed by intra-planar injection of CFA, and observed the changes of mechanical pain threshold, the number of Fos/Rab11a double-labeled cells and the expression level of Rab11a in the SDH at different time points after CFA injection. In the second part, Rab11a-specific interference virus (Rab11a-shRNA) was injected into the left SDH to observe the change of pain threshold of the ipsilateral hind paw. Combined with the first part, it was found that Rab11a positive cells were mainly NeuN positive cells, so the electrophysiological activity of these neurons in SDH after Rab11a knockdown was observed by the whole cell patch clamp technique. Thirdly, the possible mechanism of Rab11a involved in the development of chronic inflammatory pain was further explored by Immuno-Electron Microscopy and whole-cell patch clamp.
Generation of inflammatory pain model
Chronic inflammatory pain model, has often been performed to investigate the potential mechanisms of peripheral pain disorders, was constructed as our previous studies [24, 25]. After anesthetized with 2% isoflurane, 50% CFA (10 µl, Sigma-Aldrich, Saint Louis, USA) was intra-plantar subcutaneously injected into the left hindpaw of mice. Meanwhile, 0.9% saline (10 µl) was used in the control animals.
Von-Frey test
Pain withdrawal mechanical threshold (PWMT) of the left hind paw was tested via a range of Von-Frey filaments (Stoelting Company, Wood Dale, USA) as previous studies reported [26, 27]. Briefly, mice were placed in metal mesh grids (7 × 7 × 10 cm3) for 30 min before MPWT tests, which were applied at the same time point on baseline (days 0), days 1–7 after CFA/saline treatment. After mice adapted, an array of filaments (0.008, 0.02, 0.04, 0.16, 0.4, 0.6, 1, and 1.4 g) with increasing strengths (0.078, 0.196, 0.392, 1.568, 3.92, 5.88, 9.8, and 13.72 mN) were applied vertically investigate the mechanonociceptive threshold of the hind paw. The minimal bending force of filaments able to evoke 3 withdrawal response among 5 stimulations was considered the MPWT.
Immunofluorescent histochemical staining
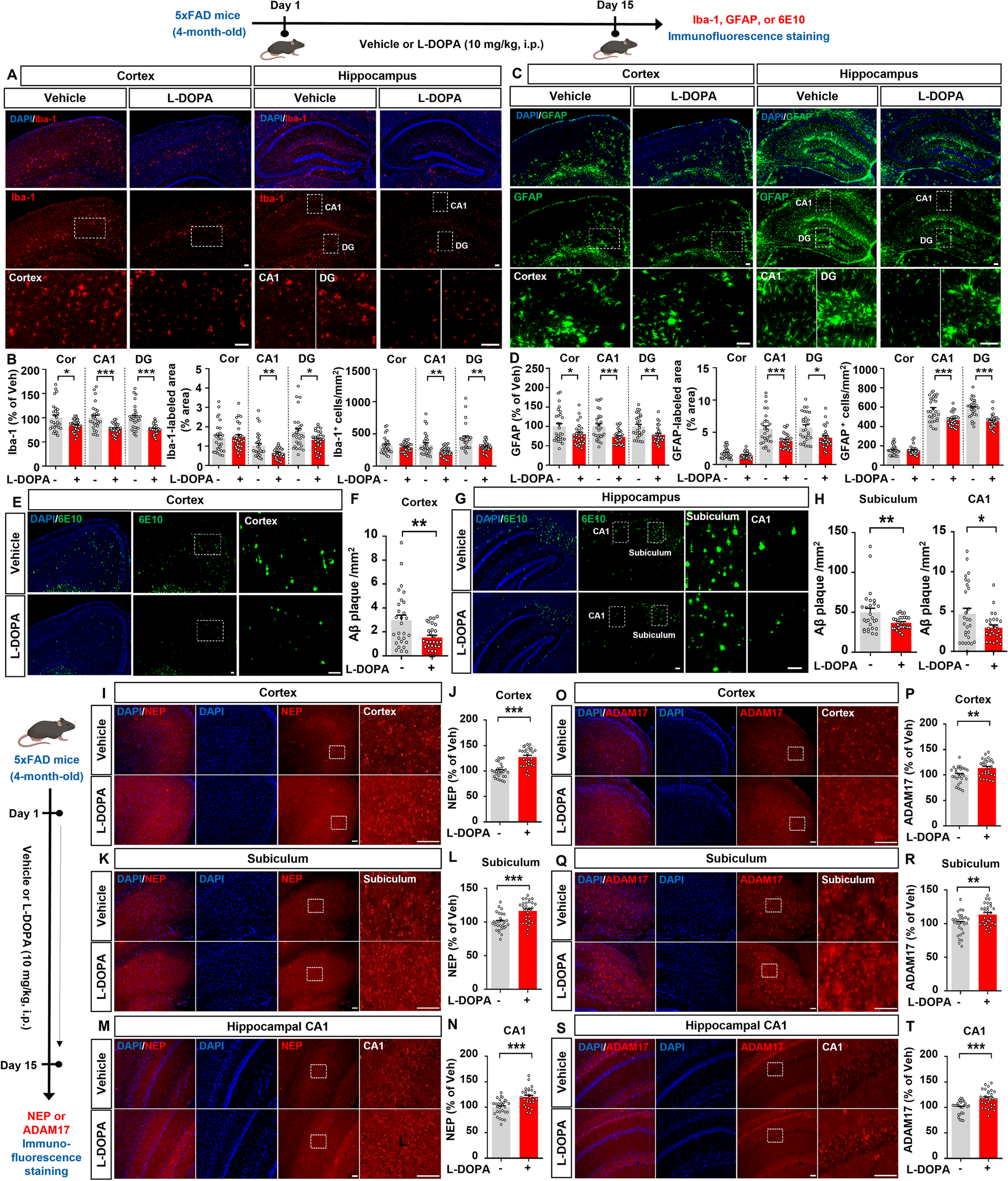
The mice were deeply anesthetized with 2% isoflurane and subsequently perfused transcardially with 25 ml of 0.01 M phosphate-buffered saline (PBS, pH 7.4), followed immediately by perfusion with 100 ml of a solution containing 4% (w/v) paraformaldehyde and 75% (v/v) saturated picric acid in 0.1 M phosphate buffer (PB, pH 7.4). The lumbar spinal cord was then rapidly removed and placed in 0.1 M PB containing 30% (w/v) sucrose overnight at 4℃. Subsequently, the spinal cord was cut into 30 μm thick serial sections using a freezing microtome (Kryostat 1720; Leitz, Mannheim, Germany). The sections were washed with 0.01 M PBS and immersed in PBS containing 0.3% Triton X-100 and 1% normal goat serum (NGS) for 30 min. All antibodies were diluted in PBS containing 5% (v/v) normal donkey serum (NDS), 0.3% (v/v) Triton X-100, 0.05% (w/v) sodium azide, and 0.25% (w/v) carrageenan (PBS-NDS, pH 7.4). For Rab11a/GFAP/Iba-1/NeuN immunofluorescent staining, the sections were incubated overnight at 4℃ with rabbit anti-Rab11a (1:200; 20229-1-AP, Proteintech, Chicago, USA), mouse anti-GFAP (1:4000; MAB3402, merckmillipore, Massachusetts, USA), mouse anti-NeuN (1:500; MAB377, merckmillipore), and goat anti-Iba-1 (1:200; ab5076, Abcam, Cambridge, UK). For Rab11a and FOS immunofluorescent staining, the sections were incubated with rabbit anti-Rab11a and mouse anti-FOS (1:500; ab11959, Abcam, Cambridge, UK) for 24 h at 4℃. After incubation with primary antibodies, the sections were washed and incubated with the appropriate fluorophore-conjugated secondary antibodies (1:200; Invitrogen, ThermoFisher, CA, USA) for 4 h at room temperature. Subsequently, the sections were washed with 0.01 M PBS three times for 10 min each. Finally, the sections were mounted on glass slides and observed using a laser scanning confocal microscope (FV1000, Olympus, Japan). The fluorescence intensity and number of double-labeled cells were measured using Image J software (NIH, Frederick, MD, USA), following the methodology described in our recent study [14].
Western blotting
The mice were deeply anesthetized with 2% isoflurane and then perfused transcardially with 25 ml of pre-cooled 0.01 M PBS (pH 7.4). The left SDHs were promptly isolated and placed in centrifuge tubes on ice. Subsequently, the left SDHs were homogenized using a hand-held pestle in sodium dodecyl sulfate (SDS) sample buffer. The samples were heated at 100 ℃ for 10 min and loaded onto 10% SDS-polyacrylamide gels using standard Laemmli solutions (BioRad Laboratories, CA, USA). Electrophoresis was performed to separate the proteins, followed by electroblotting onto a polyvinylidene difluoride membrane (PVDF, Immobilon-P, Millipore, Hayward, CA, USA). The membranes were incubated in a blocking solution for 1 h and then gently agitated overnight with rabbit anti-Rab11a (1:200). The primary antibodies bound to the membrane were detected using a horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody (1:5000; ZB-2301, ZSGB-BIO, Beijing, China). All reactions were visualized using the enhanced chemiluminescence (ECL) detection method. The densities of the protein bands were analyzed using Labworks Software (Ultra-Violet Products, UK).
Injection of viral Vector
Mice were anesthetized with 2% isoflurane and secured on a stereotaxic apparatus (RWD Life Science, Shenzhen, China). Following established protocols [7], we performed the injection procedures. To downregulate Rab11a messenger RNA (mRNA) expression, we utilized Rab11a-specific small hairpin RNA (shRNA) encoded with adeno-associated viral (AAV) vectors (rAAV-CMV-DIO-(mCherry-U6)-shRNA (Rab11a)-WPRE-hGH polyA; BrainVTA, Wuhan, China) [15]. The AAV vectors containing Rab11a-shRNA were injected into the ipsilateral SDH after co-administration with the hSyn promoter virus carrying the cre enzyme (rAAV-hSyn-CRE-WPRE-hGH pA; BrainVTA). A similar injection procedure was followed for the administration of scramble shRNA as a control. In the second part of this study, the mice were randomly divided into four groups: Sham + scramble group, Sham + shRNA group, CFA + scramble group, and CFA + shRNA group. In the Sham + scramble group and CFA + scramble group, 0.2 µl of scramble shRNA virus was injected. In the Sham + shRNA group and CFA + shRNA group, 0.2 µl of Rab11a shRNA virus was injected ipsilaterally.
Electrophysiological Recording
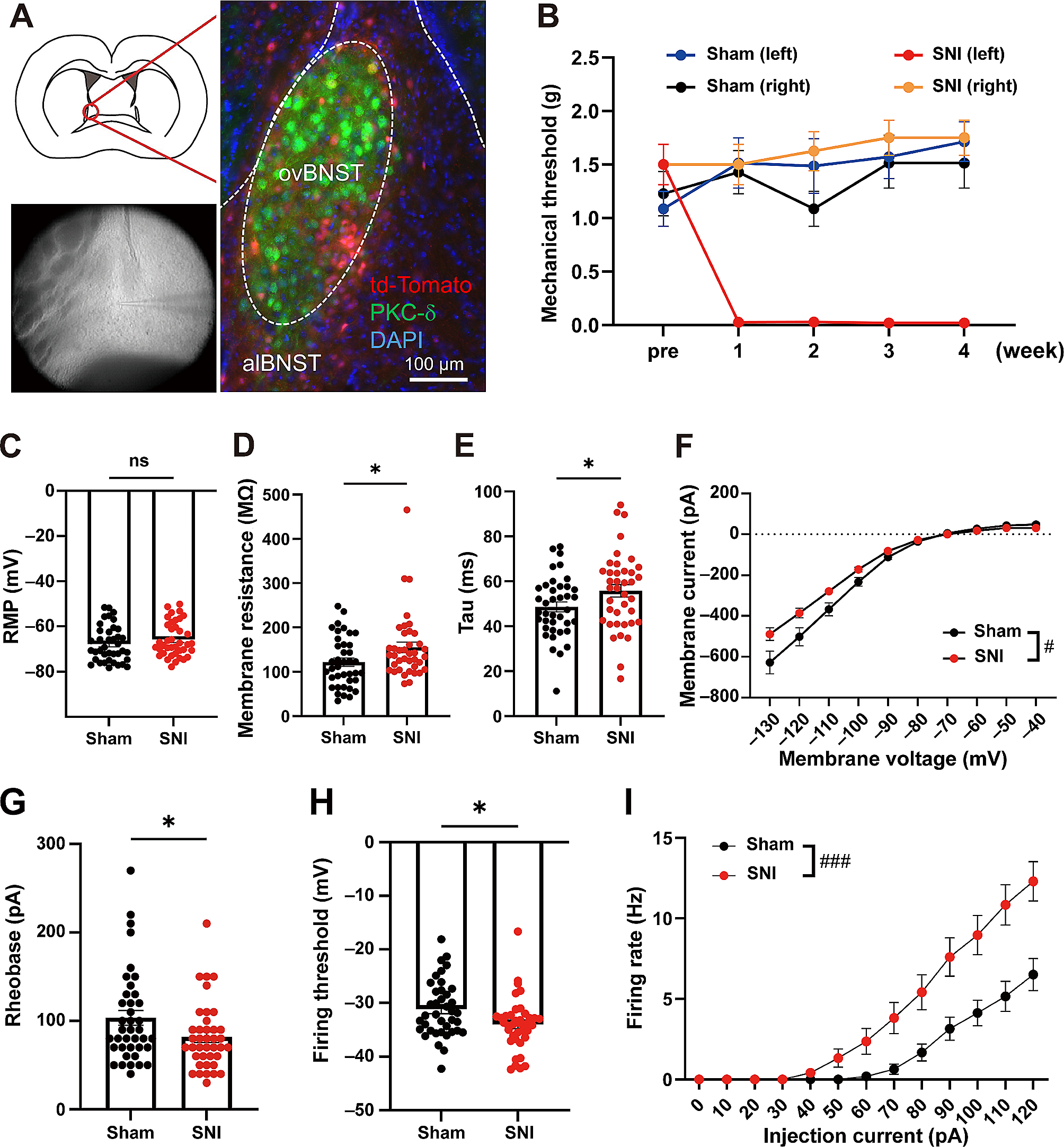
To investigate the neurophysiological properties of spinal dorsal horn (SDH) neurons, electrophysiological recordings were performed following our recent studies. On the 7th day after CFA or Sham operation, mice in the Sham + scramble group, Sham + shRNA group, CFA + scramble group, and CFA + shRNA group were anesthetized with 2% isoflurane. Lumbar spinal cord sections were then obtained and placed in oxygenated (95% O2 plus 5% CO2) pre-cooled artificial cerebrospinal fluid (ACSF) containing 248 mM sucrose instead of NaCl for 30 min at 4℃. Subsequently, 300 μm slices of the lumbar spinal cord were cut using a vibratome (Leica VT 1200 s, Heidelberger, Nussloch, Germany) and transferred into frozen oxygenated ACSF containing 124 mM NaCl, 1 mM NaH2PO4, 25 mM NaHCO3, 2 mM MgSO4·7H2O, 2.5 mM KCl, 25 mM glucose, 2 mM CaCl2, 3.0 mM pyruvate, and 1 mM ascorbate. The slices were allowed to recover at room temperature for 1 h before electrophysiological recording.
In voltage clamp mode, currents were recorded at a holding potential of -70 mV using recording pipettes filled with an intra-electrode solution consisting of 0.2 mM Tris-GTP, 0.4 mM EGTA, 4 mM Mg-ATP, 5 mM NaCl, 10 mM HEPES, 20 mM KCl, and 130 mM potassium gluconate (pH 7.2–7.4; osmolality 290–300 mOsm). Spontaneous excitatory postsynaptic currents (EPSCs) were recorded from layer I and layer II neurons using an Axon 700B amplifier (Molecular Devices Inc., CA, USA). AMPAR-mediated EPSCs were induced by repetitive stimulations at 0.02 Hz, and the neurons were voltage-clamped at -70 mV. For recording NMDA receptor-mediated EPSCs, local stimulations were delivered with a bipolar tungsten stimulating electrode connected to an isolation current stimulator [Natus Medical Incorporated (NASDAQ: PCLN-news, L6H5S1, Canada)] at an intensity of 20 µA. The recording pipettes were filled with a solution containing 102 mM cesium gluconate, 5 mM TEA chloride, 3.7 mM NaCl, 11 mM BAPTA [1,2-bis(2-aminophenoxy) ethane-N,N,N’,N’-tetraacetic acid], 0.2 mM EGTA, 20 mM Hepes, 2 mM MgATP, 0.3 mM NaGTP, and 5 mM QX-314 chloride (adjusted to pH 7.2 with CsOH, 280 to 300 mOsm). NMDA EPSCs were pharmacologically isolated in ACSF containing CNQX (6-cyano-7-nitroquinoxaline-2,3-dione; 20 mM). Neurons were voltage-clamped at + 30 mV, and NMDA EPSCs were evoked at 0.05 Hz. In current clamp mode, the firing patterns of SDH neurons in mice experiencing inflammatory pain (IP) and control mice were obtained by recording the trains of action potentials (APs) evoked by intracellular injection of depolarizing currents ranging from 0 to 200 pA (interval: 25 pA) for 400 ms.
Statistical analysis
All data in the present study are presented as mean ± standard error of the mean (SEM). Statistical analysis and data visualization were performed using GraphPad Prism 9.1 software (GraphPad Software, Inc., La Jolla, CA, USA). ImageJ software was used to analyze the density of immunofluorescence (IF) and Western blotting (WB) bands. Statistical significance was assessed using One-Way ANOVA followed by Tukey’s post-hoc test and Two-Way ANOVA followed by Tukey’s post-hoc test. All experiments were conducted in a blinded manner to minimize bias.




留言 (0)