
記住我

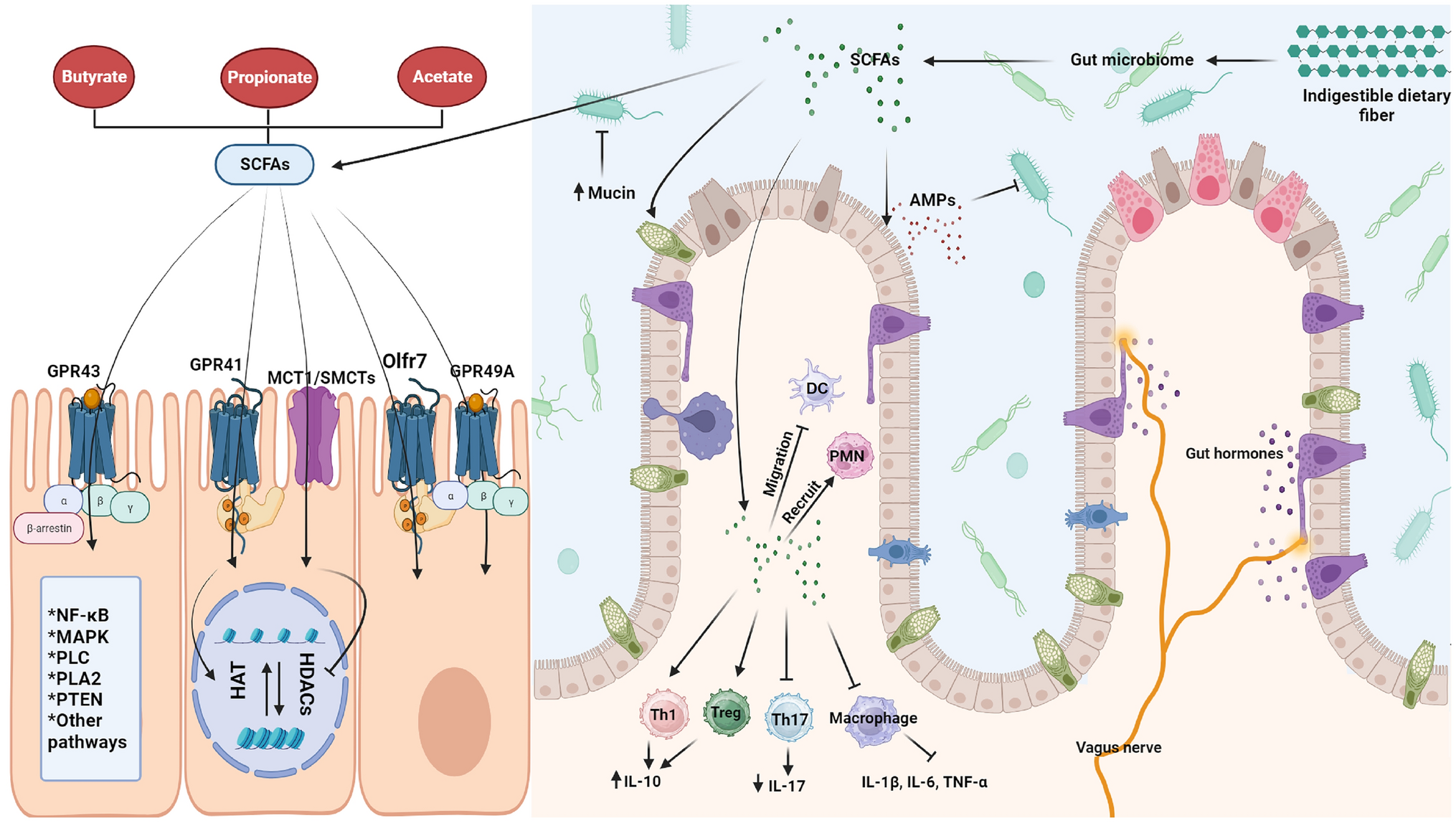

The ASD patient described here showed a mutation in the FGFR2 gene, located in the chromosomal band 10q26 (Fig. 1a) and encoding the fibroblast growth factor receptor type 2. It belongs to the family of tyrosine kinase receptors (including FGFR1, FGFR3, and FGFR4) that regulate several biological processes, including bone development, differentiation of mesenchymal and neuroectodermal cells, intracellular survival and proliferative cellular mechanisms (Azoury et al., 2017; Goyal et al., 2021). FGFR2 has been associated with several heterogeneous autosomal-dominant syndromes, among which the Pfeiffer (#MIM 101600), Crouzon (#MIM 123500), Apert (#MIM 101200) syndromes, variably presenting overlapping features affecting the skin and the skeletal and neurological systems, and often associated with a high risk of cancer. Recently, FGFR2 has been hypothesized to be implicated in neurodevelopmental disorders as well (Coci et al., 2017; Gracia-Darder et al., 2023; Szczurkowska et al., 2018; Tammimies et al., 2015).
Autosomal-dominant FGFR2 mutations might result in constitutive activation of the receptor, which, in turn, may determine an early differentiation of the osteoprogenitors, hence, a premature suture fusion (Azoury et al., 2017). Therefore, it is not surprising that most FGFR2 pathogenic variants (mainly missense ones) lead to syndromic craniosynostosis (i.e., Crouzon, Apert, Pfeiffer, Beare-Stevenson cutis gyrate, Jackson-Weiss, and Seathre-Chotzen-like syndromes) with a multisystemic involvement (Azoury et al., 2017). However, FGFR2 is nearly ubiquitously expressed, particularly in the brain, respiratory system, male and female tissues, and skin (Fig. 2); thus, mutations in this gene have been associated with other clinical phenotypes, such as Hypospadias and Lacrimo-auriculo-dento-digital syndrome (Azoury et al., 2017; Stenson et al., 2020).
Fig. 2
Expression level of the FGFR2 gene in human healthy tissues. The expression level was obtained in 54 human tissues from GTEx RNA-seq of 17,382 samples from 948 donors (V8, Aug 2019). TPM: Transcripts per Million. Data and image from the UCSC Genome Browser (http://genome.ucsc.edu, Accessed 05 May 2023)
Although the evidence available today is still sparse, recent data unravelled a potential role of FGFR2 on neurodevelopment, such as the reported presumed loss-of-function mutation (c.A1295G) of FGFR2 in an autistic child, without features of craniosynostosis (Tammimies et al., 2015). Accordingly, Gracia-Darder et al. (2023) reported a heterozygous variant (p.Cys382Arg) in FGFR2, suggesting a possible causative role in the aetiology of their patient’s autism. In addition, Coci et al. (2017) reported three siblings affected by two distinct forms of severe neuropsychological impairments inclusive of autistic features, who turned out to carry two different unbalanced translocations between chromosomes 10 and 22, deriving from a maternal balanced translocation, two chromosomes harbour important genes implicated in central nervous system development and body growth, including the FGFR2 gene. These data are summarized in Table 1.
Table 1 Summary of the clinical features of the patient here described, and comparison with cases from the literature presenting a potential involvement in Autism Spectrum Disorder of the FGFR2 geneSzczurkowska et al. (2018) have already proposed such a role for FGFR2 in neurodevelopmental disorders. They demonstrated that the mutual regulation of FGFR2 and the cell adhesion molecule NEGR1 are apparently implicated in the genesis of impaired core behaviours related to ASD in mice: the downregulation of either one of these two genes leads to a defective NEGR1-FGFR2 complex (which in its turn would converge on the Extracellular signal-regulated kinases/Akt downstream signalling pathway), eventually affecting neuronal migration and the spine density during mouse cortical development, being, therefore, responsible for the autistic symptoms. Earlier, Vaccarino et al. (2009), considering that the FGF family is involved in cortical size and connectivity regulation, mini-column pathology and excessive network excitability, hypothesized that mutations in these genes may lead to autistic sensory hyper-reactivity.
Finally, it is worth recalling our patient’s clinical features, partially overlapping with those present in specific FGFR2-related syndromes, such as high-arched palate (as in Pfeiffer syndrome), syndactyly of the 2nd and 3rd toes (Jackson-Weiss syndrome #MIM 123150 and Sathre-Chotzen syndrome, #MIM 101400), plagiocephaly (Sathre-Chotzen syndrome), developmental delay, and intellectual disability (Beare-Stevenson cutis gyrate syndrome, #MIM 123790, Crouzon syndrome, and Pfeiffer syndrome).
All these data not only corroborate the already proposed pathogenicity of FGFR2 in neurodevelopmental disorders, including autism and ADHD but also specifically support its etiological role in our patient’s case. Given the recent breakthrough of NGS techniques, such as WES, we reckon that the analysis of FGFR2 should be included, and its variants should be taken in mind in patients with neurodevelopmental disorder, especially in association with craniosynostosis and body overgrowth. Further evidence is needed to reveal the specific role of certain FGFR2 variants in determining distinct phenotypes to finally delineate a more precise genotype–phenotype correlation within this rare and intriguing group of disorders.
留言 (0)