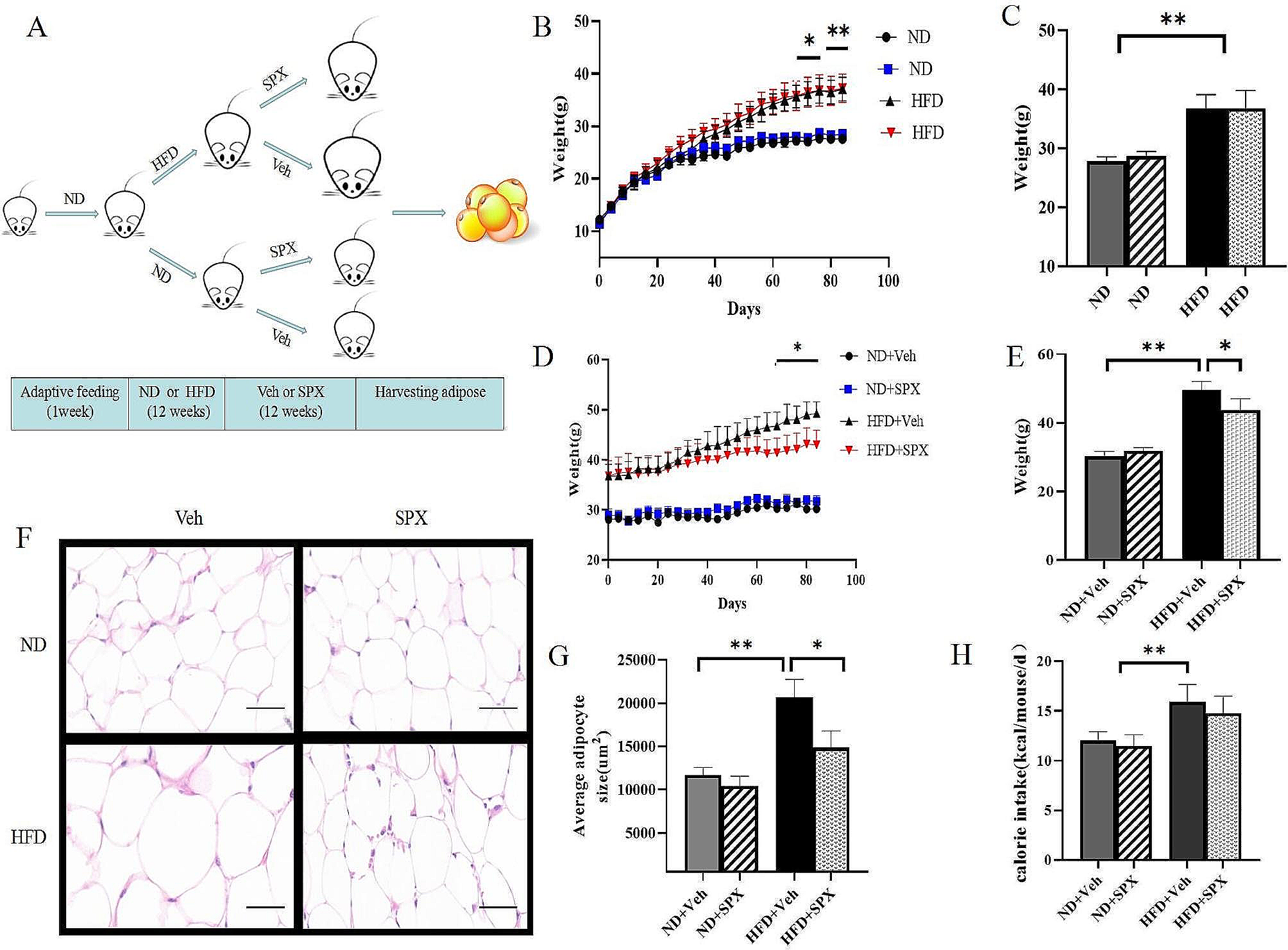
Establishment of a mouse liver IR model
C57/BL6 mice were purchased from the Experimental Animal Center of Shandong University. Mice (15 per group) were randomly divided into a test group fed on a HFD (Catalog No. D12492, Research Diets, New Brunswick) and a control group fed on a basal diet (Catalog No. D12450B, Research Diets, New Brunswick). Body weight was measured once a week and recorded. After 16 weeks, intraperitoneal insulin tolerance test (IPITT) and intraperitoneal glucose tolerance test (IPGTT) were performed, and mice in the experimental group that had become obese and developed IR (according to their blood sugar levels) were selected. The mice were dissected and their livers were used for subsequent experiments, such as detection using a gene chip, HE staining, and immunohistochemistry. All animal experiments were conducted according to protocols approved by Ethics Committee of Shandong Provincial Qianfoshan Hospital.
Dot blotting
The RNA was diluted into three different concentrations of 200 ng/μL, 100 ng/μL and 50 ng/μL. The RNA was heated for 5 min at 72 °C to denature the RNA and disrupt the secondary mechanism. The nucleic acids were fixed by aspirating drops of each concentration onto Hybond-N membranes and cross-linked twice (1200J, 25–50s) with UV 2400UV. Membrane 1 was incubated in methylene blue for 2–5 min and washed with water for 7–10 times to visualize Actin RNA. Membrane 2 was incubated in 10 mL of 5% milk for 60 min in a shaker at room temperature. The membrane was washed for 5 min three times. m6A primary antibody (Catalog No.202003, Synaptic Systems, Gottingen, Germany) was incubated overnight at 4 °C. The m6A primary antibody was recovered and the membrane was washed for 5 min three times. The secondary antibody was incubated for 60 min and the membrane was washed for 5 min three times. Western blotting luminescent solution was incubated for 5 min protected from light and then exposed.
Mouse m6A-mRNA epitranscriptomic microarray
The HFD-fed group and control group were analyzed, with three liver samples in each group. A mouse m6A epitranscriptomic microarray test (8 × 60K, Arraystar) was conducted in KangChen Biotech (No. G4102A, Shanghai, China). Generally, the sample preparation and microarray hybridization were performed based on the Arraystar’s standard protocols. The total RNAs were immunoprecipitated with anti-m6A antibody. Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images. Differentially expressed m6A methylated RNAs or differentially expressed RNAs between two comparison groups were identified by filtering with the fold change and statistical significance (p value) thresholds. Hierarchical Clustering was performed to show the distinguishable m6A methylation or expression pattern among samples.
Establishment of a hepatocyte steatosis model in HepG2 cells
The human hepatic cell line HepG2 was purchased from Procell Life Science & Technology Co., Ltd (Wuhan; China). Treatment with a mixture of FFAs (palmitic acid:oleic acid (PA:OA) = 2:1) (No. SYSJ-KJ006, Kun Chuang Technology, Xian; China) at 0, 0.25, 0.5, 0.75, 1, and 1.5 mM for 24 h was used to induce steatosis in HepG2 cells. After 48 h, according to the semiquantitative oil red O staining results, the optimal concentration to induce steatosis (1.0 mM) was selected and applied in subsequent experiments.
Semi-quantitation via oil red O staining
Observation of the HepG2 cells in which a change in fat accumulation was successfully induced under a microscope revealed obvious lipid droplet formation. These cells were fixed with tissue fixative for 10 min, after which a working solution of oil red O was added and incubated for 40 min at room temperature while avoiding light, and the sample was decolorized with a 60% isopropanol decolorizing solution for 1 min to remove background color. The cells were placed under an inverted phase-contrast microscope for observation and photography. After pictures were taken, the cells were washed twice with PBS, 100% isopropanol was added for decolorization, and the cells were mixed with the isopropanol for 10 min on a horizontal shaker. The decolorizing solution was added to a 96-well plate, with each sample was added to four subwells, and the absorbance at 510 nm was measured with a spectrophotometer.
Glucose consumption and glucose uptake
Normal HepG2 cells and FFA-induced HepG2 cells were treated as described previously. METTL3 siRNA was transfected as normal. At the end of treatment, cells were incubated with low-glucose 10% FBS-DMEM with 10–7 mol/L insulin for 4h, whereas no insulin stimulation cells were used as negative control and empty culture medium was used as blank control. For glucose consumption, glucose concentrations in the cultured medium were determined simultaneously based on hexokinase method in Cobas8000 c701 (Roche Diagnostics, Shanghai, China). After blank, the difference of glucose levels in different groups represented the glucose consumption levels. For the glucose uptake test, we conducted as described previously [19].
Cell transfections based on METTL3 and CYP2B6 interference or overexpression
METTL3 and CYP2B6 small interfering RNA (siRNA) was purchased from Gene Pharma (Wuhan; China). A negative control (NC) siRNA group was set up. The following siRNA sequences were used:
METTL3-Homo gene Sense: 5'-CCUGUAAGUAUGUUCACUATT-3', Antisense: 5'-UAGUGAACAUACUUGCAGGTT-3'.
CYP2B6-Homo gene Sense: 5'-GCGGAAUUGUUCCUCUUCUTT-3', Antisense: 5'-AGAAGAGGAACAAUUCCGCTT-3'.
Negative control (NC) Sense: 5'-UUCUCCGAACGUGUCACGUTT-3', Antisense: 5'-ACGUGACACGUUCGGAGAATT-3'.
At the same time, METTL3 and CYP2B6 were overexpressed by LV5 lentivirus transfection and with the pcDNA3.1 plasmid, respectively (GeneChem, Shanghai; China). For the transfection experiment, Lipofectamine 2000 reagent (No. 52887, Invitrogen) was applied to promote the transfection efficiency. In 12-well plates used to culture HepG2 cells, 100 μL of Opti-MEM was mixed with 2 μL of Lipofectamine 2000 and incubated at room temperature for 5 min, after which the Lipofectamine 2000 mixture was mixed with 100 μL of Opti-MEM and 4 μL of siRNA at room temperature. After 20 min, the Lipofectamine 2000 and siRNA mixture was added to 1 ml of RPMI 1640 culture medium per well. After 6–8 h, the culture medium was changed to normal medium. After 24 h of culturing, RNA was harvested and extracted and after 48 h of culturing, protein was harvested and extracted.
Extraction of total protein
Total protein was extracted using radioimmunoprecipitation assay (RIPA) lysis buffer containing protease and phosphatase inhibitors according to the manufacturer’s instructions. The cell suspension with added lysate was vortexed vigorously for 40 min, and the vortexed cell suspension was placed in a low-temperature centrifuge at 12,000 rpm for 15 min. The supernatant was aspirated into a new tube for subsequent protein quantification. Quick Start Bradford 1 × Dye Reagent was used (No. 5000205, Bio-Rad, USA) for protein quantification. Before use, the Bradford reagent was filtered. Different amounts of bovine serum albumin standard (BSA) were added to 1 mL of Bradford reagent to prepare standard protein samples at 0, 2, 4, 6, 8, and 16 µg/μL. The protein sample was added to 1 mL of the Bradford reagent and mixed well. The standard curve was independently drawn, and the protein samples to be tested were moved into 96-well plates for measurement of the absorbance at 590 nm. The concentration of the protein in each sample was calculated according to the value of the standard curve.
Extraction of membrane protein
A membrane protein extraction kit (Catalog No.89842, ThermoScientific) was applied to obtain the membrane protein. A total of 5 × 106 cells were collected and centrifuged at 1000 × g for 5 min, and the supernatant was then removed. A total of 400 μL of permeabilization buffer was added and incubate at 4 °C for 10 min (while still mixing), after which the sample was centrifuged at 4 °C and 16,000 × g for 15 min. The supernatant containing the cytoplasmic proteins was then transferred to a new tube to which 200 μL of solubilization buffer was added. The mixture was then pipetted and incubated at 4 °C for 30 min (mixing was continued) before centrifugation at 4 °C and 16,000 × g for 15 min. The supernatant containing the membrane proteins was then transferred to a new tube. The Bradford assay was used for protein quantification.
Western blotting
Samples containing 15-25μg of protein were resolved on SDS-PAGE gels, transferred to Hybond P PVDF membranes, and incubated with primary antibodies overnight at 4 °C. The anti-CYP2B6 antibody (ab140609, Abcam, Cambridge, MA) was diluted1:5000; antibodies against METTL3 (Catalog No. 96391, CST, Danvers, MA), METTL14 (Catalog No. 69159, CST, Danvers, MA), VIRILIZER (Catalog No. 69159, CST, Danvers, MA), FTO (Catalog No. 45980, CST, Danvers, MA), ALKBH5 (Catalog No. 80283, CST, Danvers, MA), WTAP (Catalog No. 69159, CST, Danvers, MA), ERK (Catalog No. 4377, CST, Danvers, MA), AKT (Catalog No. 4691, CST, Danvers, MA), phosphatidylinositol 3-kinase (PI3K) (Catalog No. 4292, CST, Danvers, MA), IRS1 (Catalog No. AF2587, Beyotime, China), and Glut4 (Catalog No. 2213, CST, Danvers, MA) were diluted 1:1000; and anti-β-actin was diluted 1:2500. The bands containing different target proteins were incubated with anti-rabbit or anti-mouse secondary antibodies for 1 h at room temperature. After exposure, the differences between the strips were compared by routine laboratory protocol.
Immunohistochemistry
Liver tissues were used to make paraffin sections for immunohistochemistry experiments. The slices were placed in different concentrations alcohol for dewaxing and a 3% hydrogen peroxide solution to deactivate endogenous peroxidase to avoid overly intense staining. The slices were placed in a citrate buffer solution (0.01M) in a pressure cooker and heated at medium–high heat for antigen repair and after 2 min the slices were cooled to room temperature. Normal goat serum was used to block the slices for 1 h to prevent non-specific binding of the antibodies to tissues. Primary antibody anti-Cyp2b10 (Catalog No. sc-73546, Santa Cruz, CA, 1:200) was added and incubated overnight at 4 °C. Secondary antibody was added and incubated at room temperature for 1 h. Diaminobenzidine was then added to promote color development, hematoxylin was used to counterstain the nuclei, and 0.5% hydrochloric acid in alcohol was used for color separation. The slices were dehydrated, mounted on slides, and observed under a microscope.
RT-PCR and real-time PCR
Total RNA was extracted by the TRIzol method, and 2μL of RNA was used to determine the RNA concentration. Two micrograms of RNA were reverse transcribed using a RevertAid First Strand cDNA Synthesis Kit (Catalog No. K1622, ThermoScientific) to synthesize cDNA using the following conditions: 25 °C for 5 min, 42 °C for 60 min, and 70 °C for 5 min, followed by a holding step at 4 °C. APowerUp SYBR Green Master Mix Kit (No. A25742, ThermoScientific) was used to perform real-time PCR. The sequence of each synthetic primer is shown in (Table 1).
Table 1 The synthetic sequence information of primers used for RT-PCRMeRIP-qPCR
A total of 2 × 107 cells were collected and RNA was extracted by the TRIzol method. A 1% agarose gel was prepared and the quality of the RNA was measured by electrophoresis. A methylated RNA immunoprecipitation (MeRIP) kit was purchased from Bersinbio Biotechnology (Catalog No. Bes5203, Guangzhou, China). Fragmentation buffer was added to the complete RNA and the mixture was heated at 94 °C for 1 min to fragment the RNA into 100-nt fragments. m6A-specific antibody or IgG was added to the fragmented RNA and the mixture was incubated at 4 °C for 2 h to allow the RNA fragments to bind to the antibody or IgG. Magnetic beads were simultaneously blocked for 2 h. The blocked magnetic beads were mixed with the RNA fragments bound to the antibody; more specifically, the magnetic beads bound to the RNA fragments which were bound to the anti-m6A antibody. Proteinase K was used to digest the magnetic beads for 45 min and the supernatant was transferred to a new tube. The RNA was extracted, subjected to RNA quantification and reverse transcription, and used for subsequent real-time PCR experiments. Sequence information of m6A specific primers used for MERIP-qPCR was shown in (Table 2).
Table 2 Sequence information ofm6A sites specific primers used for MERIP-qPCRDetection of RNA m6A modifications based on LC–MS/MS
RNA m6A modifications contents were detected by MetWare (http://www.metware.cn/) based on the AB Sciex QTRAP 6500 LC–MS/MS platform. Generally, sample preparation and nucleosides extraction were conducted based on generally protocol. After RNA was digested into nucleosides completely, the mixture was extracted with chloroform. The resulting aqueous layer was collected for analysis with LC-ESI-MS/MS. The sample extracts were analyzed using an UPLC-ESI-MS/MS system based on optimized conditions. The service was completed by METVIARE (Wuhan, China).
STM2457 injection experiment in vivo
C57/BL6 male mice (eight per group) were randomly divided into four groups: basal CD group, HFD + 0.9% NaCl injection group, HFD + DMSO injection group, and HFD + STM2457 (Catalog No. 2499663-01-1, Selleck Chemicals, Houston, Texas) injection group. When the mean body weight of mice in HFD group was statistically different from that of mice in the control group [(mean body weight in HFD—mean body weight in CD)/mean body weight in CD > 30%], the treatment was started once a day for one or two weeks. STM2457 was administered at a dose of 50 mg/kg and weighed prior to each injection. DMSO or saline was also administered at an equal volume. Body weight was recorded every day. Injection of STM2457 was conducted daily and totally for one or two weeks.
Determination of liver TC and TG in mice
Livers from different groups of mice were collected and homogenized fully. TC and TG levels were assayed and compared based on the protocol for assay lab kits for TC (Catalog No. E-BC-K109-M, Elabscience, Wuhan, China) and TG (Catalog No. E-BC-K261-M, Elabscience, Wuhan, China).
Data analyses
The data are presented as the mean ± standard error of the mean (SEM). Comparisons between the means of two groups were analyzed using a t test. One-way analysis of variance (ANOVA) was performed when more than two groups were compared using the SPSS 16.0 software package. A q test was used for further pairwise comparisons. P values of less than 0.05 were considered to indicate statistical significance.



留言 (0)