
記住我
NEC is the most common and fatal gastrointestinal emergency in the neonatal intensive care unit (NICU) (1, 2), and 90% of these infants who develop NEC are preterm (3, 4). Epidemiological results show that the total incidence of NEC in NICU infants is 2% to 5%, among which the morbidity and mortality very low birth weight (VLBW; <1,500 g) neonates are 4.5%−8.7% and 20%−30%, respectively, while the mortality rate in extremely low birth weight (ELBW; <1,000 g) infants is even as high as 30% to 50.9% (5–7). A better understanding of the pathogenesis of NEC is the key to finding better diagnostic, preventive, and therapeutic strategies.
The intestinal tract of premature infants is characterized by incomplete development of immune defense function, and impairment of the intestinal epithelial barrier (IEB) function exacerbated after experiencing hypoxia and hypothermia (8, 9). The disruption of IEB is a key factor in the development of intestinal inflammation (10), while increased intestinal permeability caused by IEB injury can be further amplified by intestinal inflammation, thus forming a positive feedback loop (10). Furthermore, the aggravated intestinal permeability and altered intestinal microbiome work together to cause the transfer of bacteria or bacterial products across the IEB, triggering a more intense inflammatory response, and even adverse consequences such as NEC, late-onset sepsis (LOS), intestinal perforation, and death (11–13). Therefore, it has been proposed that NEC evolves from the destruction of the IEB (14) and further develops into LOS in the imbalance between IEC damage and repair (15).
The anti-permeability of IEB is mainly maintained and regulated by intestinal epithelial cells (IECs) and the tight junction (TJ) formed between IECs in the apical region (16). The intestinal epithelium is a single-cell layer structure composed of multiple cells differentiated from intestinal stem cells (ISCs) (17). Eighty percent of the IECs are absorptive villi epithelial cells, which are responsible for maintaining the barrier function of the IEB, while the Paneth cells located in the basement of the intestinal crypts are considered to be “reserve ISCs” and play an important role in the repair of the intestinal epithelium (18). Severe injury of IEB and increased intestinal permeability are the most prevalent microscopic phenotypes in NEC (19). More importantly, increased intestinal permeability occurs before the appearance of NEC signs (20), and the destruction of IEB in turn makes premature infants susceptible to NEC (21). Therefore, understanding the cellular damage mechanism of IECs is key to revealing IEB dysfunction.
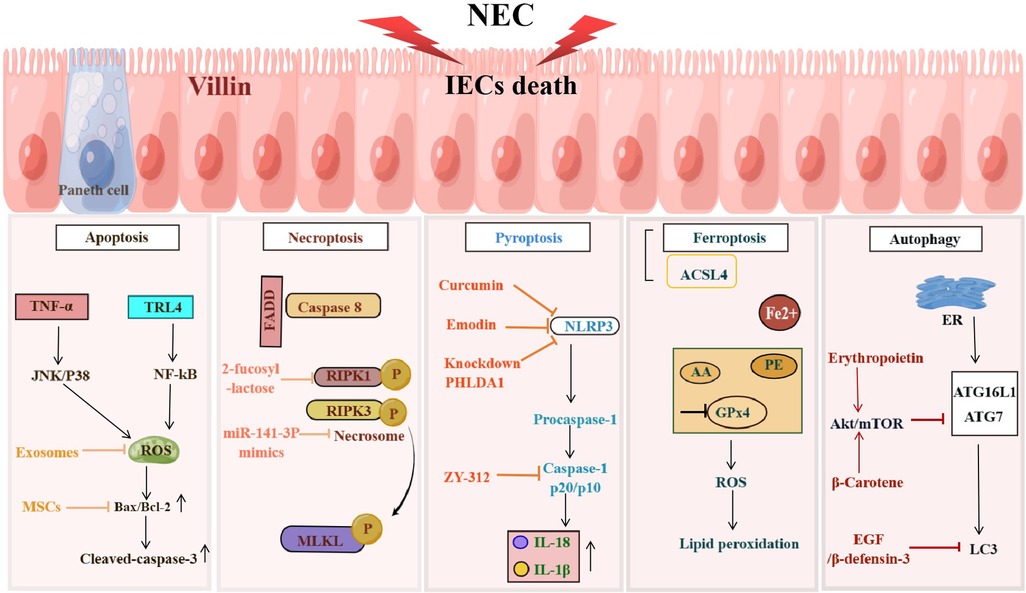
2. Death of IECsThe definition of NEC is derived from the fact that IECs die in this disease (22). Therefore, one of the most basic problems in the field of NEC research is how do IECs die. Answering this question is essential for elucidating the pathogenesis of NEC. The currently known death modes of IECs in NEC mainly include apoptosis, necroptosis, pyroptosis, ferroptosis, and abnormal autophagy (Figure 1).
Figure 1. NEC induces programmed death of IECs. Programmed cell death including apoptosis, necroptosis, pyroptosis, ferroptosis and autophagy occurs in IECs during the development of NEC, resulting in the damage to the IEB. The figure shows the signaling pathways in different death types of IECs.
2.1. IECs apoptosisIntestinal epithelial cells have an extremely high rate of self-renewal, and their self-clearance at the end of the life cycle in an apoptotic manner is essential. However, excessive apoptosis may exacerbate the disruption of the intestinal barrier, allowing more microorganisms to enter the submucosa and producing large amounts of cytokines, and further inducing IECs apoptosis (23). For example, TNF-α leads to apoptosis of IECs by inducing mitochondrial reactive oxygen species production and activating the JNK/p38 signaling pathway (24). In the neonatal rat NEC model, the mRNA ratio of Bax/Bcl-2 and the protein expression level of Bax were significantly increased in the damaged ileum tissue. Besides, the number of cleaved-caspase-3 positive cells was also markedly increased in the epithelial cells of the terminal ileum. Considering that the ileum is the most vulnerable intestinal tissue in NEC, these results indicate the existence of abnormal apoptosis of IECs in NEC (25).
In the small intestinal tissue of neonatal mice, TLR4 activates the NF-κB signaling pathway and leads to increased apoptosis of intestinal cells (26). In contrast, LPS-induced IECs apoptosis and the incidence of NEC were markedly reduced in TLR4 knockout mice (27). In addition, TLR4-induced endoplasmic reticulum stress also led to increased apoptosis of ISCs (28), thereby affecting the repair of the intestinal epithelium. An increased mRNA ratio of Bax/Bcl-2 was also observed in the resected ileal tissues from NEC infants (29). A large number of TUNEL-positive cells were observed in the apical villus cells, and these cells also showed positive nitrotyrosine staining, suggesting that the apoptosis of IECs may also be associated with the formation of peroxynitrite caused by NO (30).
Despite species differences, human breast milk-derived exosomes have been shown to prevent H2O2-induced cytotoxicity and reduce oxidative stress damage in IECs in a mouse NEC model (31). Oxidative stress is one of the main factors that increase the expression of the tumor suppressor p53, which promotes apoptosis (32). Breast milk-derived exosomes were further shown to regulate IECs injury by targeting p53 through the delivery of miRNA-125b (33).
In addition, stem cell therapy has been gradually applied in NEC treatment in recent years. Stem cells have self-renewal ability, multi-directional differentiation, and good applicability. Injecting stem cells into NEC model rats can effectively inhibit apoptosis, reduce inflammation and protect the intestinal barrier (34, 35). Mesenchymal stem cells (MSCs) have been shown to reduce the incidence and severity of experimental NEC in rats (36, 37). Intraperitoneal injection of amniotic fluid-derived mesenchymal stem cells (AF-MSCs) can suppress Bax expression by activating Bib and upregulating CHOP, thereby resisting the apoptosis of IECs caused by endoplasmic reticulum stress (38, 39). Stem cell therapy has been successfully applied to clinical cases in 2019 (40), but more evidence is still needed to further elucidate the therapeutic mechanisms of stem cells in NEC to achieve breakthroughs in clinical trials.
2.2. IECs necroptosisAlthough apoptosis has long been recognized as the major mode of IECs death in NEC (25, 41). However, intestinal inflammation is an important hallmark of NEC (21), whereas apoptosis, as a non-inflammatory type of cell death (42), seems insufficient to explain the rapid and intense inflammatory response in NEC, suggesting that apoptosis is more like a consequence than a cause of NEC (22). Programmed necrosis is another highly regulated type of programmed cell death also known as necroptosis, which is a highly inflammatory type of cell death independent of caspase activation (43). Necroptosis is characterized by the formation of necrosome upon activation of receptor-interacting protein kinases (RIPK1 and RIPK3), which in turn activate mixed-lineage kinase-like (MLKL). Phosphorylated MLKL oligomerizes and translocates to the cell membrane, resulting in cell membrane rupture and release of damage-associated molecular patterns (DAMPs) to trigger an inflammatory cascade response (43). Necroptosis is associated with the pathogenesis of a variety of mucosal inflammatory diseases (44), including inflammatory bowel disease and allergic colitis in children, and lethal ileitis during intestinal development (45, 46). Recent studies have reported that necroptosis is observed in IECs of human and mouse NEC models, especially in differentiated villous epithelial cells. Blocking necroptosis by inhibitors or gene knockdown can reduce the pathological damage of IEB in NEC mice (22), suggesting that necroptosis of IECs plays an important role in NEC-related IEB injury. Interestingly, in addition to mediating apoptosis of IECs, TLR4 also mediates necroptosis in the development of NEC (47).
The cell types of IECs affected by apoptosis and necroptosis are different in the pathogenesis of NEC. Apoptosis occurs mainly in the intestinal crypt epithelial cells (e.g., Paneth cells) (28, 33), while necroptosis mainly occurs in intestinal epithelial villi cells (22). Differences in the death types of IECs may be related to their different biological roles in the pathogenesis of NEC. Apoptosis of ISCs can lead to intestinal mucosal epithelial repair disorder, affecting the recovery of IEB function (29). Necroptosis explains the rupture of intestinal villi in NEC, which facilitates microbial translocation across the IEB and promotes the development of LOS (22). In addition, necroptosis that occurs in infiltrating lymphocytes in the neonatal intestine can further promote the loss of IECs (22). Therefore, necroptosis of IECs may be a more direct pathological death mode in IEB lesions due to NEC.
2-fucosyl lactose isolated from breast milk can effectively prevent the activation of necroptosis-characteristic genes in the ileum tissue of premature infants with NEC (22). This result partially explains why infants fed breast milk have a lower risk of NEC than infants fed formula milk, which is due to the lack of 2-fucose lactose in formula milk. miR-141-3p mimics can inhibit the up-regulation of necroptosis-related molecules and the interaction of RIPK1 and RIPK3 in LPS-induced Caco-2 cells (48), suggesting that miR-141-3p protects IECs from cellular injury by inhibiting RIPK1-mediated necroptosis and inflammatory cytokine release, providing a perspective for exploring the pathogenesis of NEC.
2.3. IECs pyroptosisPyroptosis is another inflammatory mode of programmed cell death reported in recent years (49). It mainly relies on inflammasomes to activate caspase-1, resulting in the shearing of gasdermin D (GSDMD). Subsequently, the cleaved GSDMA translocates to the cell membrane and forms pores through polymerization, leading to cell membrane rupture and cell death, accompanied by the release of large amounts of cytokines (50). In addition, activated caspase-1 also cleaves the precursors of IL-1β and IL-18 to form mature IL-1β and IL-18, which are then released into the extracellular and aggravate the inflammatory response (51). Cronobacter sakazakii has been reported to induce NEC. The underlying mechanism is to promote the activation of NLRP3 inflammasome by activating TLR4/MyD88/NF-kB signaling pathway, resulting in the up-regulation of downstream caspase-1 expression and the increase of IL-1β release, and ultimately inducing pyroptosis of intestinal cells (52). Significantly increased mRNA expression of IL-1β, IL-18 and NLRP3, and protein expression of caspase-1 p20/p10 were also observed in the ileocecal region of NEC model mice (53). Therefore, there seems to be the possibility of intestinal epithelium pyroptosis in the animal models of NEC, but more detailed pathological diagnosis and anti-pyroptosis treatment studies still need to be further carried out. In addition, increased levels of IL-1β were detected in the peripheral blood of infants with NEC, and the expression of IL-1β and caspase-1 was also observed in the intestinal epithelium (54).
Curcumin attenuates NLRP3/caspase-1-induced pyroptosis by activating the SIRT1/NRF2 signaling pathway or inhibiting TLR4 expression, thereby reducing intestinal injury in the rat model of NEC (55). Emodin has also been reported to reduce intestinal pyroptosis in NEC neonatal rats by inhibiting the NLRP3/IL-1β pathway (56). Recently, PHLDA1 was found to be highly expressed in the intestinal tissues of NEC mice. In sh-PHLDA1 transfected mice with NEC, the survival rate was increased, and intestinal inflammation, oxidative stress, and cell scorching were also improved significantly (57). Knockdown of PHLDA1 was shown to alleviate the NEC phenotype by activating Nrf2 to inhibit NLRP3 activation and pyroptosis (57). Fan et al. isolated a novel probiotic candidate strain from fecal samples of healthy infants and characterized it as Bacteroides fragilis strain ZY-312 (58). ZY-312 can reduce Cronobacter sakazakii-induced pyroptosis by inhibiting caspase-1 and decreasing IL-1β in NEC rats. Interestingly, ZY-312 also reversed the increased levels of caspase-3 and Bax/Bcl-2, suggesting that ZY-312 could simultaneously block apoptosis induced by Cronobacter sakazakii (58). As a promising probiotic agent, ZY-312 has great utility for the prevention and treatment of NEC by reducing dual cell death forms (pyroptosis and apoptosis) to restore IEB function and alleviate local inflammatory response.
2.4. IECs ferroptosisFerroptosis is an iron-dependent programmed cell necrosis characterized by lipid peroxidation and membrane damage (59). In recent years, several studies have demonstrated the presence of ferroptosis in IEB pathological injury in a variety of colorectal diseases including ulcerative colitis (60, 61), Crohn's disease (62), and intestinal ischemia/reperfusion injury (63, 64). Ferroptosis leads to elevated levels of lipid peroxidation (LPO)by inhibiting the activation of glutathione peroxidase 4 (GPx4) and the oxidation of arachidonic acid (AA) and its esterified phosphatidylethanolamine (PE) (63, 65), resulting in the death of IECs and disruption of the intestinal mechanical barrier (66). Antioxidant enzyme activity is lower in neonates compared to adults, making infants more susceptible to oxidative stress (67). Peroxidation-induced ROS accumulation is thought to produce a strong pro-inflammatory effect in NEC-associated ferroptosis (68). Bioinformatic analysis and wet experiments have demonstrated the involvement of ferroptosis in NEC and found that ACSL4, a key regulator of ferroptosis execution, may participate in NEC by activating NEC-related Toll-like receptor signaling pathway to induce IECs death and immune cell activation (69). Meanwhile, ACSL4 is also involved in autophagy, pyroptosis, apoptosis, hypoxia, and inflammation of NEC (69). However, there is still a lack of anti-ferroptosis therapy in NEC. There is currently little evidence to support the ferroptosis of IECs in NEC, and the specific mechanism needs to be further confirmed, which will also provide new insights into the treatment of NEC.
2.5. IECs autophagyAutophagy is a process of lysosome-dependent degradation of proteins and organelles and interacts with apoptosis to synergistically eliminate aging, redundant or damaged cells (70). Excessive autophagy has been identified as a risk factor for NEC development (71). Hackam et al. previously reported that gene expression levels of TLR4, ATG7, LC3, ATG16 and Beclin1 were significantly increased in the fetal intestine compared to full-term infants, and that intestinal tissues obtained from aborted fetuses also showed significantly increased staining for LC3 within the intestinal epithelium (72). Human β-defensin 3 (hBD3) treatment inhibits excessive autophagy in IECs may through the CXCR4 signaling pathway. Meanwhile, hBD3 significantly reduces the expression of autophagy-related proteins (Beclin1, LC3 and p62) and inflammatory cytokines, leading to improved migration of IECs and intestinal mucosal integrity, as well as a reduction in mortality in NEC rats (73). Similarly, erythropoietin in breast milk protects IECs from excessive autophagy and apoptosis in NEC mice via the Akt/mTOR and MAPK/ERK signaling pathway (74). Epidermal growth factor (EGF) treatment attenuates NEC injury by modulating intestinal autophagy in rat models (71, 75). β-carotene also attenuates LPS-induced apoptosis and autophagy in IECs by activating the PI3K/AKT/mTOR signaling pathway (76). All these studies suggest that IECs autophagy and apoptosis play an important role in NEC pathological damage, and modulating IECs autophagy can be used as an effective complement to anti-apoptotic therapy for NEC.
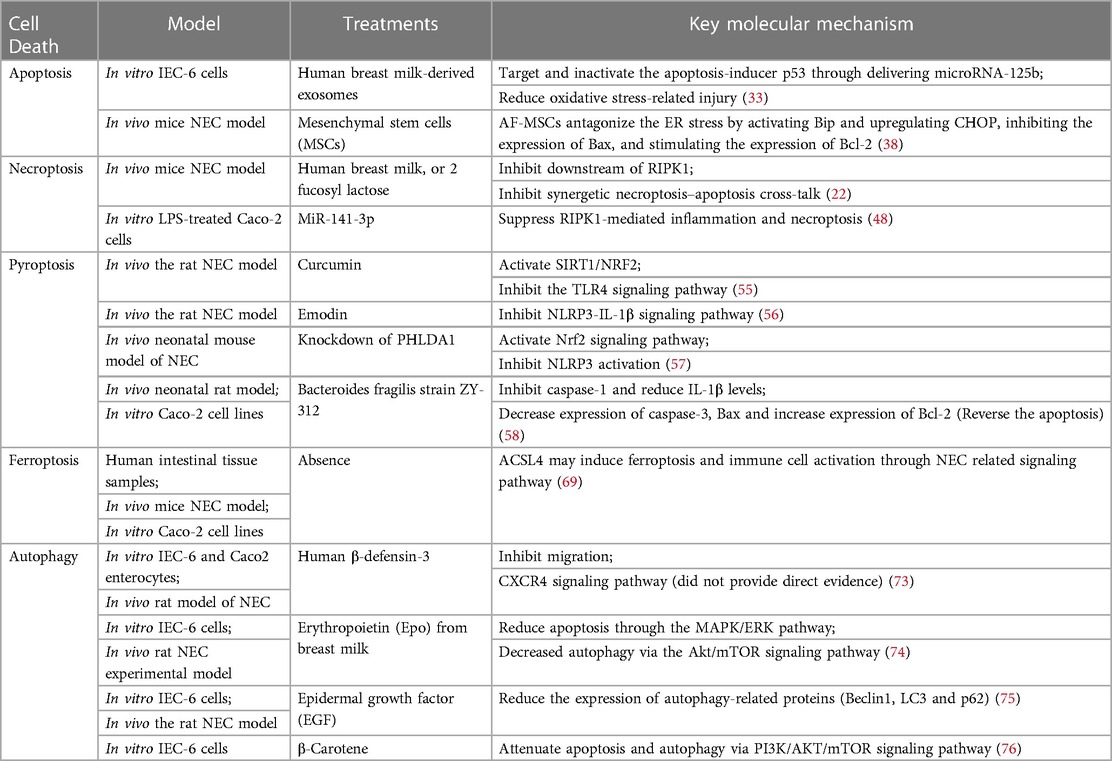
3. Conclusion and outlookHow does the pathological IECs death mechanism interact and induce increased intestinal permeability, leading to intestinal barrier dysfunction? Can targeting one of these cell death mechanisms as a therapeutic strategy provide effective treatment for infants with NEC? Although these problems remain questionable, understanding the mechanisms of IECs' death has brought new insights to explore new treatments (77). In this review, we highlight the important role of IECs death mechanisms in the process of NEC and review potential therapeutic approaches to alleviate NEC by preventing IECs death in recent years (Table 1). Future breakthroughs in NEC treatment should focus on finding drugs or interventions that can effectively protect IEB. Mechanistically, it is desirable that these drugs directly target the programmed death of IECs and the associated severe IEB damage caused by inflammation.
Table 1. Therapies and drugs targeting different IECs death pathway.
Author contributionsZW and TY conceptualized; ZW and SY drafted the manuscript; XW, YZ, CG and XL performed the literature collation; ZW, YT and XW reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
FundingThis work was supported by grants from the National Natural Science Foundation of China (82104269, 81872880), the Science and Technology Commission of Shanghai Municipality (21140905300), and Shanghai “Rising Stars of Medical Talents” Youth Development Program-Youth Medical Talents: Clinical Pharmacist Program (SHWSRS (2021) _099).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References4. Stoll BJ, Hansen NI, Bell EF, Walsh MC, Carlo WA, Shankaran S, et al. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993-2012. Jama. (2015) 314(10):1039–51. doi: 10.1001/jama.2015.10244
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Jiang S, Yan W, Li S, Zhang L, Zhang Y, Shah PS, et al. Mortality and morbidity in infants <34 Weeks’ gestation in 25 NICUs in China: a prospective cohort study. Front Pediatr. (2020) 8:33. doi: 10.3389/fped.2020.00033
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Eaton S, Rees CM, Hall NJ. Current research on the epidemiology, pathogenesis, and management of necrotizing enterocolitis. Neonatology. (2017) 111(4):423–30. doi: 10.1159/000458462
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Moore SA, Nighot P, Reyes C, Rawat M, McKee J, Lemon D, et al. Intestinal barrier dysfunction in human necrotizing enterocolitis. J Pediatr Surg. (2016) 51(12):1907–13. doi: 10.1016/j.jpedsurg.2016.09.011
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Tanner SM, Berryhill TF, Ellenburg JL, Jilling T, Cleveland DS, Lorenz RG, et al. Pathogenesis of necrotizing enterocolitis: modeling the innate immune response. Am J Pathol. (2015) 185(1):4–16. doi: 10.1016/j.ajpath.2014.08.028
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Denning TL, Bhatia AM, Kane AF, Patel RM, Denning PW. Pathogenesis of NEC: role of the innate and adaptive immune response. Semin Perinatol. (2017) 41(1):15–28. doi: 10.1053/j.semperi.2016.09.014
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Nanthakumar N, Meng D, Goldstein AM, Zhu W, Lu L, Uauy R, et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS One. (2011) 6(3):e17776. doi: 10.1371/journal.pone.0017776
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Hackam DJ, Upperman JS, Grishin A, Ford HR. Disordered enterocyte signaling and intestinal barrier dysfunction in the pathogenesis of necrotizing enterocolitis. Semin Pediatr Surg. (2005) 14(1):49–57. doi: 10.1053/j.sempedsurg.2004.10.025
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Chokshi NK, Guner YS, Hunter CJ, Upperman JS, Grishin A, Ford HR. The role of nitric oxide in intestinal epithelial injury and restitution in neonatal necrotizing enterocolitis. Semin Perinatol. (2008) 32(2):92–9. doi: 10.1053/j.semperi.2008.01.002
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Capaldo CT, Powell DN, Kalman D. Layered defense: how mucus and tight junctions seal the intestinal barrier. J Mol Med (Berl). (2017) 95(9):927–34. doi: 10.1007/s00109-017-1557-x
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Kandasamy J, Huda S, Ambalavanan N, Jilling T. Inflammatory signals that regulate intestinal epithelial renewal, differentiation, migration and cell death: implications for necrotizing enterocolitis. Pathophysiology. (2014) 21(1):67–80. doi: 10.1016/j.pathophys.2014.01.001
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, et al. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature. (2013) 495(7439):65–9. doi: 10.1038/nature11965
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Martin CR, Walker WA. Intestinal immune defences and the inflammatory response in necrotising enterocolitis. Semin Fetal Neonatal Med. (2006) 11(5):369–77. doi: 10.1016/j.siny.2006.03.002
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Bergmann KR, Liu SX, Tian R, Kushnir A, Turner JR, Li HL, et al. Bifidobacteria stabilize claudins at tight junctions and prevent intestinal barrier dysfunction in mouse necrotizing enterocolitis. Am J Pathol. (2013) 182(5):1595–606. doi: 10.1016/j.ajpath.2013.01.013
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Halpern MD, Denning PW. The role of intestinal epithelial barrier function in the development of NEC. Tissue Barriers. (2015) 3(1-2):e1000707. doi: 10.1080/21688370.2014.1000707
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Werts AD, Fulton WB, Ladd MR, Saad-Eldin A, Chen YX, Kovler ML, et al. A novel role for necroptosis in the pathogenesis of necrotizing enterocolitis. Cell Mol Gastroenterol Hepatol. (2020) 9(3):403–23. doi: 10.1016/j.jcmgh.2019.11.002
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Caplan MS, Simon D, Jilling T. The role of PAF, TLR, and the inflammatory response in neonatal necrotizing enterocolitis. Semin Pediatr Surg. (2005) 14(3):145–51. doi: 10.1053/j.sempedsurg.2005.05.002
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Baregamian N, Song J, Bailey CE, Papaconstantinou J, Evers BM, Chung DH. Tumor necrosis factor-alpha and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy, and c-Jun N-terminal kinase/p38 phosphorylation during necrotizing enterocolitis. Oxid Med Cell Longev. (2009) 2(5):297–306. doi: 10.4161/oxim.2.5.9541
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Clark JA, Lane RH, Maclennan NK, Holubec H, Dvorakova K, Halpern MD, et al. Epidermal growth factor reduces intestinal apoptosis in an experimental model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. (2005) 288(4):G755–62. doi: 10.1152/ajpgi.00172.2004
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Richardson WM, Sodhi CP, Russo A, Siggers RH, Afrazi A, Gribar SC, et al. Nucleotide-binding oligomerization domain-2 inhibits toll-like receptor-4 signaling in the intestinal epithelium. Gastroenterology. (2010) 139(3):904–17. 917.e1-6. doi: 10.1053/j.gastro.2010.05.038
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Leaphart CL, Cavallo J, Gribar SC, Cetin S, Li J, Branca MF, et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol. (2007) 179(7):4808–20. doi: 10.4049/jimmunol.179.7.4808
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Afrazi A, Branca MF, Sodhi CP, Good M, Yamaguchi Y, Egan CE, et al. Toll-like receptor 4-mediated endoplasmic reticulum stress in intestinal crypts induces necrotizing enterocolitis. J Biol Chem. (2014) 289(14):9584–99. doi: 10.1074/jbc.M113.526517
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Ates U, Gollu G, Kucuk G, Billur D, Bingol-Kologlu M, Yılmaz Y, et al. Increase in pro-apoptotic Bax expression and decrease in anti-apoptotic Bcl-2 expression in newborns with necrotizing enterocolitis. Arch Argent Pediatr. (2016) 114(3):243–7. doi: 10.5546/aap.2016.eng.243
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Ford H, Watkins S, Reblock K, Rowe M. The role of inflammatory cytokines and nitric oxide in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg. (1997) 32(2):275–82. doi: 10.1016/S0022-3468(97)90194-9
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Martin C, Patel M, Williams S, Arora H, Brawner K, Sims B. Human breast milk-derived exosomes attenuate cell death in intestinal epithelial cells. Innate Immun. (2018) 24(5):278–84. doi: 10.1177/1753425918785715
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Simon K, Mukundan A, Dewundara S, Van Remmen H, Dombkowski AA, Cabelof DC. Transcriptional profiling of the age-related response to genotoxic stress points to differential DNA damage response with age. Mech Ageing Dev. (2009) 130(9):637–47. doi: 10.1016/j.mad.2009.07.007
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Neal MD, Sodhi CP, Jia H, Dyer M, Egan CE, Yazji I, et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J Biol Chem. (2012) 287(44):37296–308. doi: 10.1074/jbc.M112.375881
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Sajeesh S, Broekelman T, Mecham RP, Ramamurthi A. Stem cell derived extracellular vesicles for vascular elastic matrix regenerative repair. Acta Biomater. (2020) 113:267–78. doi: 10.1016/j.actbio.2020.07.002
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Pisano C, Besner GE. Potential role of stem cells in disease prevention based on a murine model of experimental necrotizing enterocolitis. J Pediatr Surg. (2019) 54(3):413–6. doi: 10.1016/j.jpedsurg.2018.07.025
PubMed Abstract | CrossRef Full Text | Google Scholar
36. McCulloh CJ, Olson JK, Zhou Y, Wang Y, Besner GE. Stem cells and necrotizing enterocolitis: a direct comparison of the efficacy of multiple types of stem cells. J Pediatr Surg. (2017) 52(6):999–1005. doi: 10.1016/j.jpedsurg.2017.03.028
PubMed Abstract | CrossRef Full Text | Google Scholar
37. McCulloh CJ, Olson JK, Wang Y, Vu J, Gartner S, Besner GE. Evaluating the efficacy of different types of stem cells in preserving gut barrier function in necrotizing enterocolitis. J Surg Res. (2017) 214:278–85. doi: 10.1016/j.jss.2017.03.026
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Li B, Lee C, Chuslip S, Lee D, Biouss G, Wu R, et al. Intestinal epithelial tight junctions and permeability can be rescued through the regulation of endoplasmic reticulum stress by amniotic fluid stem cells during necrotizing enterocolitis. Faseb J. (2021) 35(1):e21265. doi: 10.1096/fj.202001426R
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Lau E, Lee C, Li B, Pierro A. Endoplasmic reticulum stress in the acute intestinal epithelial injury of necrotizing enterocolitis. Pediatr Surg Int. (2021) 37(9):1151–60. doi: 10.1007/s00383-021-04929-8
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Akduman H, Dilli D, Ergün E, Çakmakçı E, Çelebi SK, Çitli R, et al. Successful mesenchymal stem cell application in supraventricular tachycardia-related necrotizing enterocolitis: a case report. Fetal Pediatr Pathol. (2021) 40(3):250–5. doi: 10.1080/15513815.2019.1693672
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Buret AG, Bhargava A. Modulatory mechanisms of enterocyte apoptosis by viral, bacterial and parasitic pathogens. Crit Rev Microbiol. (2014) 40(1):1–17. doi: 10.3109/1040841X.2012.746952
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, et al. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol. (2014) 109(2):279–87. doi: 10.1038/ajg.2013.403
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Shindo R, Ohmuraya M, Komazawa-Sakon S, Miyake S, Deguchi Y, Yamazaki S, et al. Necroptosis of intestinal epithelial cells induces type 3 innate lymphoid cell-dependent lethal ileitis. iScience. (2019) 15:536–51. doi: 10.1016/j.isci.2019.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Liu T, Zong H, Chen X, Li S, Liu Z, Cui X, et al. Toll-like receptor 4-mediated necroptosis in the development of necrotizing enterocolitis. Pediatr Res. (2022) 91(1):73–82. doi: 10.1038/s41390-021-01457-y
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Li X, Wang Y, Wang Y, He X. MiR-141-3p ameliorates RIPK1-mediated necroptosis of intestinal epithelial cells in necrotizing enterocolitis. Aging (Albany NY). (2020) 12(18):18073–83. doi: 10.18632/aging.103608
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. (2017) 277(1):61–75. doi: 10.1111/imr.12534
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Yang J, Liu Z, Wang C, Yang R, Rathkey JK, Pinkard OW, et al. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc Natl Acad Sci U S A. (2018) 115(26):6792–7. doi: 10.1073/pnas.1800562115
PubMed Abstract | CrossRef Full Text | Google Scholar
51. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD Membrane pore formation constitutes the mechanism of pyroptotic cell death. Embo J. (2016) 35(16):1766–78. doi: 10.15252/embj.201694696
PubMed Abstract | CrossRef Full Text | Google Scholar
52. Chen Z, Zhang Y, Lin R, Meng X, Zhao W, Shen W, et al. Cronobacter sakazakii induces necrotizing enterocolitis by regulating NLRP3 inflammasome expression via TLR4. J Med Microbiol. (2020) 69(5):748–58. doi: 10.1099/jmm.0.001181
PubMed Abstract | CrossRef Full Text | Google Scholar
53. Hu D, Liu H. Pyroptosis is involved in the pathogenesis of necrotizing enterocolitis. Xi bao yu fen zi Mian yi xue za zhi=Chinese Journal of Cellular and Molecular Immunology. (2018) 34(12):1070–4. doi: 10.13423/j.cnki.cjcmi.008718
CrossRef Full Text | Google Scholar
54. Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O'Donnell JA, et al. NLRP1 Inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. (2012) 37(6):1009–23. doi: 10.1016/j.immuni.2012.08.027
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Yin Y, Wu X, Peng B, Zou H, Li S, Wang J, et al. Curcumin improves necrotising microscopic colitis and cell pyroptosis by activating SIRT1/NRF2 and inhibiting the TLR4 signalling pathway in newborn rats. Innate Immun. (2020) 26(7):609–17. doi: 10.1177/1753425920933656
留言 (0)