
記住我
Epithelioid sarcoma (ES), a malignant mesenchymal tumor that occurs in extremities of adolescents, was originally described by Enzinger in 1970 (1). In 1997, Guillion et al. (2) identified proximal ES after observing ES in perineum, genital tract, and pelvis in young people. Recurrence and metastasis are prevalent. Clinically, ES of the adrenal is extremely rare, with a few cases having been documented. In this report, we describe an adrenal ES case with a focus on imaging findings and immunohistochemical indexes to promote disease awareness.
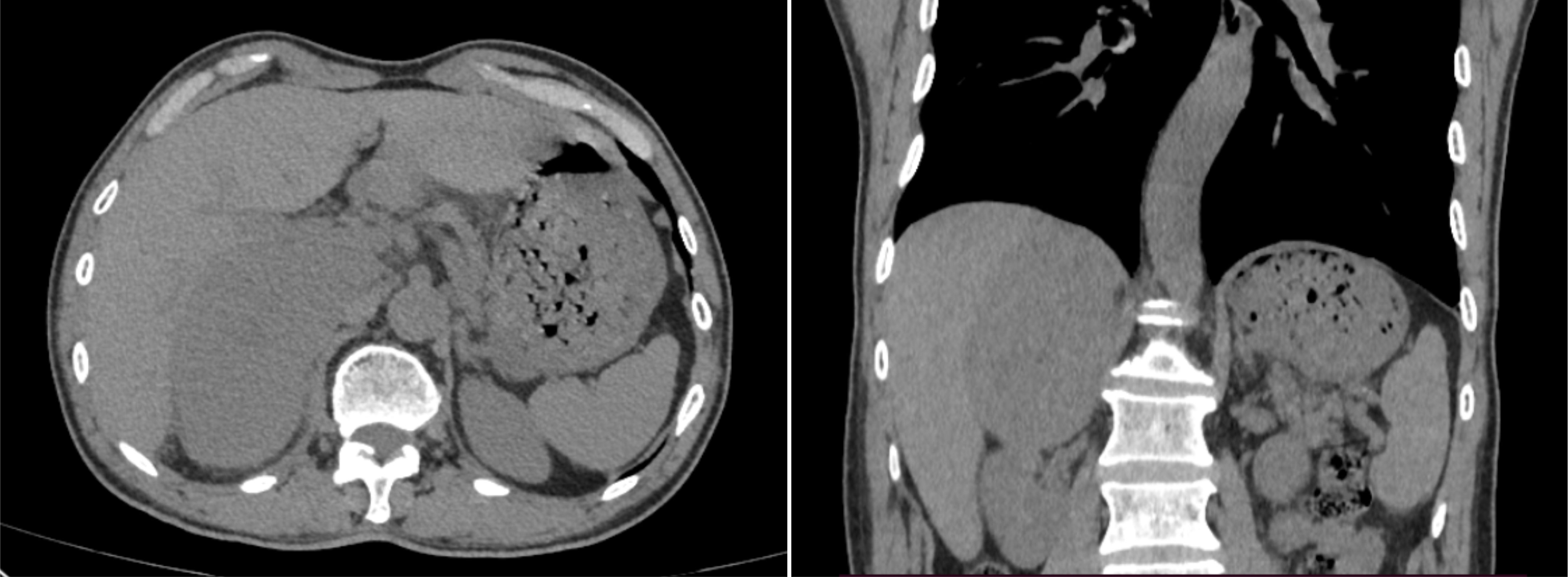
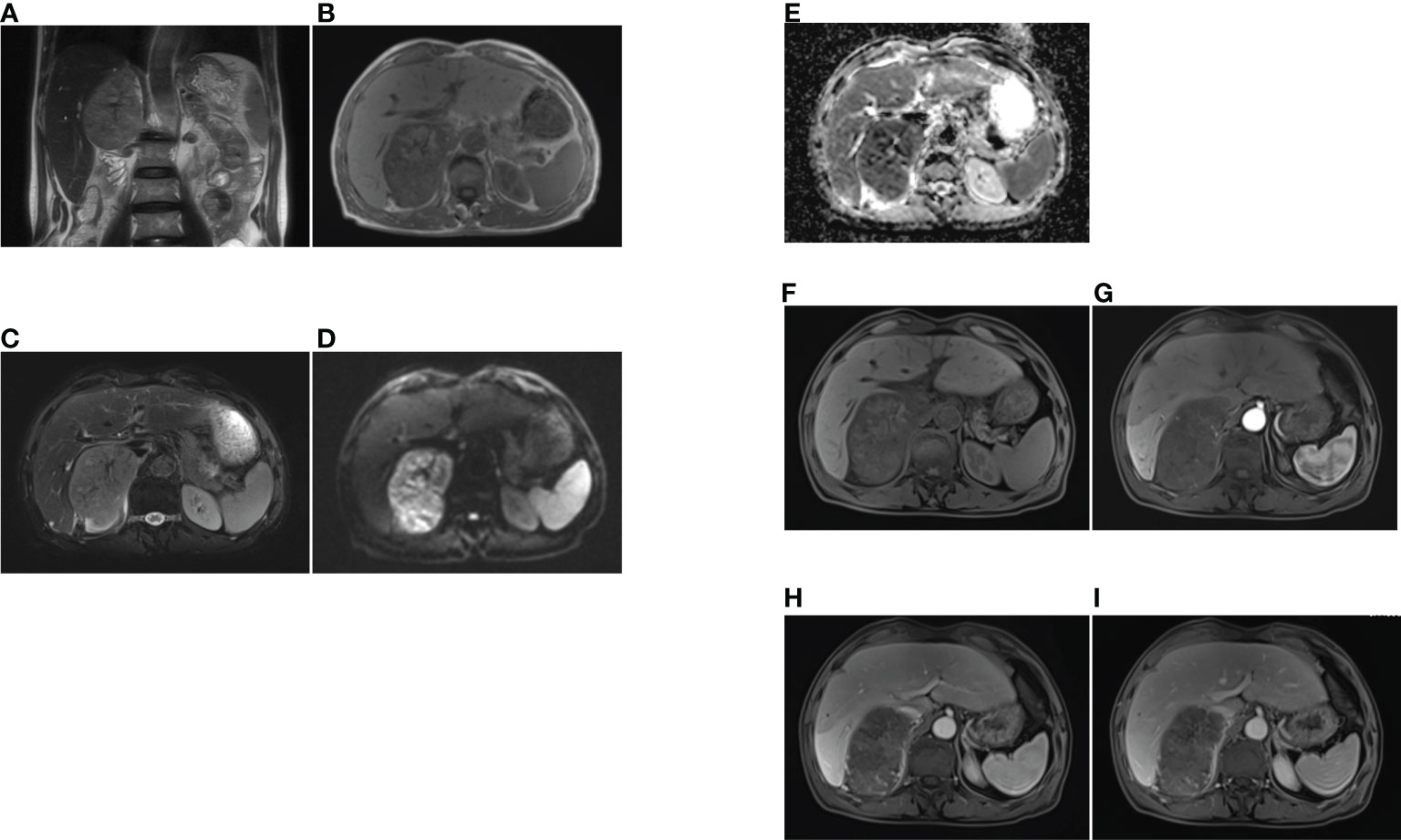
Case presentationA 69-year-old man with an incidental right adrenal gland mass was admitted to our institution. He had no relevant past medical history, and no family history was identified. Hematological examination did not reveal any abnormal findings. Adrenal function laboratory assay indices were normal, including cortisol, catecholamine and aldosterone. Initial CT scan revealed a round-like, well-defined slightly hypodense lesion in the right adrenal gland region measuring approximately 10.6×6.4×11.0 cm in size, with a CT value of about 34 HU. The right posterior liver, portal vein and upper pole of the right kidney were displayed (Figure 1). Right adrenal gland imaging was not conclusive. A follow-up MRI (Figure 2) revealed a 10.2×6.5×10.5 cm oval tumor in the right suprarenal region, with a high-low mixed sign on T1WI and slightly high signals on fat suppression T2WI sequence. A central scar was noted in the lesion, which was hypointense on T1WI and T2WI. Upon enhancement scan, the lesion exhibited a slight to moderate heterogeneous enhancement with no enhancement in the central scar. Visualization of the right adrenal gland was not clear. There were no significant enlarged lymphnodes or metastatic foci. Gene detection and FDG PET/CT were not carried out due to economic reasons.
Figure 1 Selected window images of axial, coronal plain abdomen CT scan soft tissue demonstrating a heterogeneous adrenal mass measuring 10.6×6.4×11.0cm, which abutted and compressed the liver parenchyma, right renal and IVC.
Figure 2 The MRI findings of primary adrenal sarcoma. (A) coronal T2-weighted MR image, (B) axial T1-weighted MR image, (C) axial fat suppression T2-weighted MR image, (D, E) The tumor showed heterogeneous high signal intensity on DWI and low signal intensity on ADC map, (F–I) axial fat suppression T1-weighted MR imaging (F), and Gadolinium-enhanced T1-weighted MR mages at arterial (G), portal (H) and delayed phases (I), the tumor exhibited heterogeneous enhancement and no enhancement in the central scar.
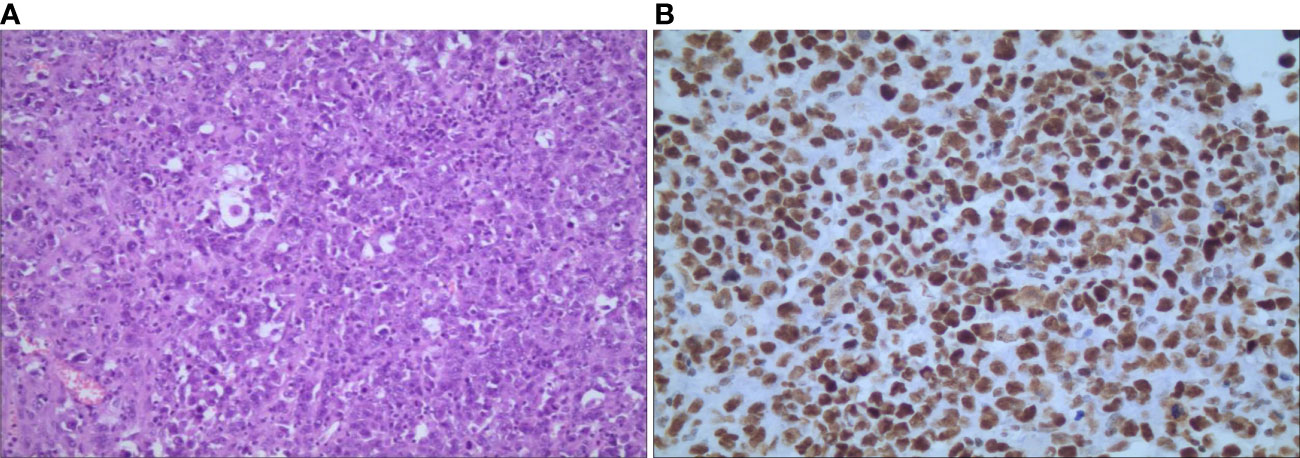
Given the extensive region of focus, laparoscopic right adrenalectomy and intestine adhesion relaxation were performed. Intraoperative findings showed that the right adrenal was occupied by a round-like mass with a diameter of about 10 cm, while the normal adrenal tissue was compressed. Due to the huge size, adhesion of the lesion and liver was obvious and the inferior vena cava was compressed. Pathological examination revealed an incomplete capsule in the adrenal tissue, whose cut surface was gray, reddish-gray and soft. Microscopically, polygonal cells were established to be distributed separately, scattered, in clusters orpatches, with large, vesicular nuclei and prominent nucleoli. Immunohistochemical analysis showed that tumor cells were positive for CK8/18, EMA, vimentin, CD10, Desmin, CD34, SMARCB1/INI-1, SOX-2, SALL4, p53 and Ki-67, while SMARCA4/BRG1, SMARCA 2(BRM), Cr, S-100, HMB-45, Pax-2, Pax-8, WT-1, CK5/6, Melan-A, CK-7, Alpha-inhibin, RCC, MyoD1, Myogenin and SMA were negative (Figure 3). Based on the above findings, the patient was diagnosed with right adrenal epithelioid sarcoma. About two months after the operation, PET/CT examination showed recurrence with multiple systemic metastases. Then, the patient underwent chemotherapy of anlotinib combined with anti-PD-1 antibody and epirubicin for 1 cycle. At present, he has a good response, showing the reduction or disappearance of tumor mass, and close follow-up is continuing.
Figure 3 Microscopic findings of primary adrenal epithelioid sarcoma. (A) hematoxylin-eosin staining shows that polygonal cells were distributed separately, scattered, in clusters orpatches, with large nuclei and prominent nucleoli (magnification, ×100); (B) immunochemical staining shows nuclear positivity for INI-1 (magnification, ×400).
Discussion and conclusionEpithelioid sarcoma (ES), a malignant soft tissue sarcoma with a certain degree of epithelization, accounts for < 1% of soft-tissue sarcomas. The two pathological subtypes of ES are classical and proximal types. The conventional type is more common and is often located in superficial dermis or subcutaneous tissues of distal limbs of young adults, whereas the proximal type is quite often located in the proximal soft tissue of elderly patients, such as the limb band, trunk and pelvis. The tumor cells of this subtype are higher grade and demonstrate a rhabaoid phenotype, compared to the classical type.
Because it is highly rare, accurate diagnosis of the primary ES of solid organs by radiologists is difficult. To elucidate on the disease, we report a case of adrenal ES, which is the fifth reported case of ES arising from the adrenal gland.
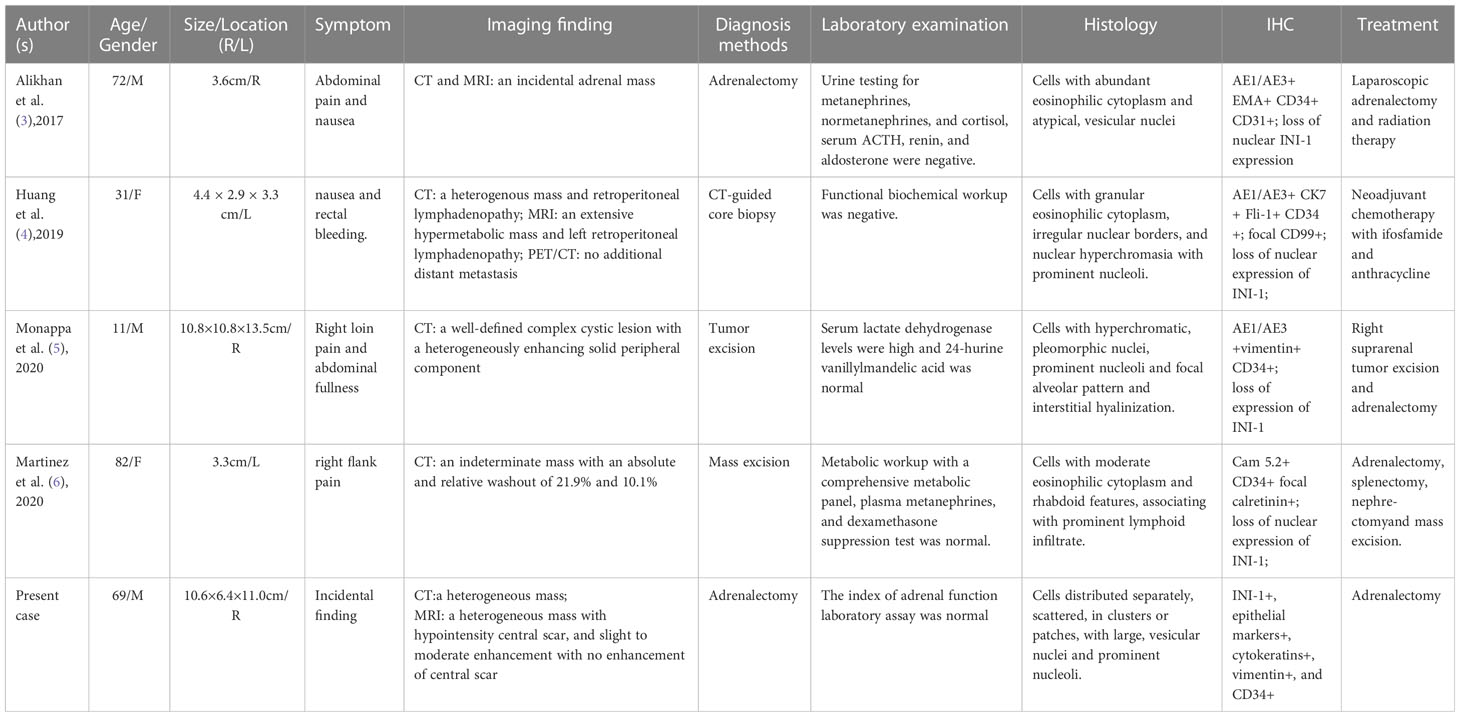
For our case, the mass was detected during physical examination, without obvious clinical manifestations. In a review of previous CT imaging in 2019, no adrenal mass was found, indicating rapid progress. Reports on MRI and CT of proximal ES involving uterine body, cervix and renal have been relatively few. To date, four cases of primary adrenal epithelioid sarcoma have been reported, but imaging features were not mentioned (3–6). A typical CT of proximal ES reveals slightly lower or equal density mass in homogeneous enhancement on enhanced scan. For the larger, manifestations include tissue invasion, hemorrhage and necrosis, but it is rarely to find calcification (7). On MRI, proximal ES is usually homogeneous in signal, isointense or hyperintense in T1WI and T2WI, and homogeneous or heterogeneous enhancement, with no enhancement in the necrotic area. There is infiltration to adjacent tissues or bones in some cases (8). In our case, plain CT revealed a slightly low-density mass in the right adrenal gland. Upon MRI, the mass exhibited heterogeneous isointensity in T1WI and relative hyperintensity with hypointensity central scar inside in fat-suppression T2WI. The mass had hyperintense signals on DWI and hypointense signals on the ADC map, while the central scar presented hypointensity. After Gd-DTPA enhancement scanning, it also showed progressive and slight to moderate enhancement on fat-suppression T1WI, with no enhancement of the central scar. The CT findings were comparable to those reported earlier. Due to the small number of reported patients, characteristic imaging findings should be investigated further for comprehensive understanding of the disease. A summary of clinical characteristics and imaging findings of the four reported cases of primary adrenal ES are listed in Table 1.
Table 1 Case reports of primary ES of adrenal gland.
Malignant lesions with adrenal masses should be considered in differential ES diagnosis. Adrenocortical adenocarcinoma, the most common primary malignant tumor of the adrenal gland, is usually characterized by irregular, huge and ill-defined mass (>5 mm in diameter), prone to central necrosis, and 20~30% with calcification. Lesion enhancement scan findings revealed a heterogeneous enhancing mass that descended and attenuated slowly (9). Angiosarcoma is a well-defined heterogeneous tumor that is characterized by bleeding and calcification. The necrosis area of angiosarcoma is greater, compared to that of ES. Meanwhile, it has specific imaging features on enhanced CT or MRI: demonstrating peripheral enhancement in arterial phase and expansion to the center in venous and delayed phase, which is similar to typical findings of hemangioma (10, 11). A large mass with intra-tumor necrosis and adjacent structure invasion are common imaging findings of leiomyosarcoma, but bleeding and calcification are rare. The solid portion is persistently enhanced, while the area of cystoid variation and necrosis are not enhanced (12). Visible tumor neovascularization and needlepoint-like blood vessels in the tumor can reflect its characteristics and be of great value for differential diagnosis. ES should also be differentiated from Sarcomatoid carcinoma, which presents as large cystic and solid mass, and evident delayed enhancement of the wall and septa (13).
In previous studies, immunohistochemical analysis of adrenal ES revealed positive expressions of epithelial markers, cytokeratins, vimentin, CD34, and negative expressions of CD31, SMARCB1/INI-1 and S-100. In particular, the loss of SMARCB1/INI-1 is considered to be the most specific aspect of immunohistochemical features for the diagnosis of ES. SMARCB1/INI-1 is one of a component of SW1/SNF chromatin-remodeling complex, which is the main condition factor for regulating gene expression (14). It is noteworthy that SMARCB/INI-1 was positive in our case, while the loss of SMARCB1/INI-1 expression has been detected in all previous reports of adrenal ES. The positive expression of SMARCB1/INI-1 often suggests malignant rhabdoid features, which may be correlated with the core subunit protein deficiency or other abnormalities causing dysfunction of SWI/SNF chromatin remodeling (15). Kohashi et al. (16) found that SMARCB1/INI-1 preserved ES predicts more aggressive biology behaviors than epithelioid sarcoma with loss of SMARCB1/INI-1 expression. Therefore, the expression of SMARCB1/INI-1 may be an important index of poor prognosis of proximal ES.
In conclusion, we report a case of proximal adrenal ES, which is rare and not commonly suspected in clinical practice. However, due to the high aggressiveness, poor prognosis, and prone to recurrence and metastasis, radiotherapy and chemotherapy after radical surgical resection is the optimal treatment. Currently, definite diagnosis of adrenal ES depends on histopathologic and immunohistochemical characteristics, but imaging examination displays some features and plays a significant role in diagnosis and differential diagnosis, providing the relationship with nearby structures. Therefore, awareness of the above may be useful for clinical diagnosis and treatment.
Data availability statementThe original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statementWritten informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributionsHY reviewed the literatures and wrote the initial draft of this manuscript. YZ prepared the images and for collected the data of the patient. LW revised the manuscript for English grammar. All authors contributed to the article and approved the submitted version. JZ supervised and reviewed the manuscript.
FundingThis work was supported by Jiangsu Province’s 333 High-Level Talent Project (grant number: BRA2020193), Jiangsu Provincial Medical Youth Talent (grant number: LGY2018032).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer (1970) 26(5):1029–41. doi: 10.1002/1097-0142(197011)26:5<1029::aid-cncr2820260510>3.0.co;2-r
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Guillou L, Wadden C, Coindre JM, Krausz T, Fletcher CD. “Proximal-type” epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol (1997) 21(2):130–46. doi: 10.1097/00000478-199702000-00002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Alikhan MB, Pease G, Watkin W, Grogan R, Krausz T, Antic T. Primary epithelioid sarcoma of the kidney and adrenal gland: report of 2 cases with immunohistochemical and molecular cytogenetic studies. Hum Pathol (2017) 61:158–63. doi: 10.1016/j.humpath.2016.09.024
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Monappa V, Singh VK, Chawla A. Primary adrenal epithelioid sarcoma in a child: a case report with literature review. Fetal Pediatr Pathol (2020) 41(1):134–40. doi: 10.1080/15513815.2020.1745972
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Tateishi U, Hasegawa T, Kusumoto M, Yokoyama R, Moriyama N. Radiologic manifestations of proximal-type epithelioid sarcoma of the soft tissues. AJR Am J Roentgenol (2002) 179(4):973–7. doi: 10.2214/ajr.179.4.1790973
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hanna SL, Kaste S, Jenkins JJ, Hewan-Lowe K, Spence JV, Gupta M, et al. Epithelioid sarcoma: clinical, MR imaging and pathologic findings. Skeletal Radiol (2002) 31(7):400–12. doi: 10.1007/s00256-002-0509-9
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Bharwani N, Rockall AG, Sahdev A, Gueorguiev M, Drake W, Grossman AB, et al. Adrenocortical carcinoma: the range of appearances on CT and MRI. AJR Am J Roentgenol (2011) 196(6):W706–14. doi: 10.2214/AJR.10.5540
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Li XM, Yang H, Reng J, Zhou P, Cheng ZZ, Li Z, et al. A case report of primary adrenal angiosarcoma as depicted on magnetic resonance imaging. Med (Baltimore) (2017) 96(45):e8551. doi: 10.1097/MD.0000000000008551
CrossRef Full Text | Google Scholar
12. Jabarkhel F, Puttonen H, Hansson L, Muth A, Ragnarsson O. Primary adrenal leiomyosarcoma: clinical, radiological, and histopathological characteristics. J Endocr Soc (2020) 4(6):a55. doi: 10.1210/jendso/bvaa055
CrossRef Full Text | Google Scholar
13. Ishikawa N, Nagase M, Takami S, Araki A, Ishikawa N, Koike C, et al. A case report of bilateral sarcomatoid carcinoma of adrenal glands with adrenal insufficiency. Int J Surg Pathol (2016) 24(8):743–8. doi: 10.1177/1066896916657589
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Kohashi K, Tanaka Y, Kishimoto H, Yamamoto H, Yamada Y, Taguchi T, et al. Reclassification of rhabdoid tumor and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 protein expression: three subtypes of rhabdoid tumor according to their histological features. Mod Pathol (2016) 29(10):1232–42. doi: 10.1038/modpathol.2016.106
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Kohashi K, Yamamoto H, Yamada Y, Kinoshita I, Taguchi T, Iwamoto Y, et al. SWI/SNF chromatin-remodeling complex status in SMARCB1/INI1-preserved epithelioid sarcoma. Am J Surg Pathol (2018) 42(3):312–8. doi: 10.1097/PAS.0000000000001011
留言 (0)