
記住我
The genus Pestivirus, belonging to the family Flaviviridae, comprises pathogens responsible for massive economic losses in livestocks, especially pigs and ruminant species (1–3) and often cause clinical manifestations ranging from mild to severe (4–7). Among pestiviruses, classical swine fever virus (CSFV) and bovine viral diarrhea virus 1 and 2 (BVDV-1 and BVDV-2) are the most impactful ones. Notably, the majority of pestiviruses are non-cytopathogenic (NCP), while both two biological types: NCP and cytopathogenic (CP) viruses have been reported in CSFV and BVDV strains isolated in clinical samples (4). Several other viruses related to pestivirus have been also described in some studies. These viruses isolated from domestic animals (8–17) and wild species (18–21) have great differences in genetics. Recently, the following eleven viruses: BVDV-1, BVDV-2, CSFV, BDV, pronghorn pestivirus, Bungowannah virus, giraffe pestivirus, HoBi-like pestivirus, Aydin-like pestivirus, rat pestivirus, and atypical porcine pestivirus have been appointed to Pestivirus A-K, respectively (3, 15).
Pestiviruses could transmit from one species of ruminants to another frequently. For example, ruminant pestiviruses often infect pigs (22). However, no evidence is available to suggest the replication of CSFV in ruminants. After infection, pestiviruses are excreted through various body secretions and usually transmitted by direct contact with infected animals or indirect contact with infectious secretions, contaminated food, or needles (23). Animals with pestiviruses infection (especially those with persistent infection) excrete lots of viruses from their body secretions for life.
Pestiviruses are single-stranded, positive-sense RNA viruses with an envelope and a genomic size of about 12.3 kb (24). The genomic RNA is translated into a single polyprotein, which is processed subsequently into four structural proteins (SPs): a basic core protein C and three envelope (E) glycoproteins Erns, E1, and E2, as well as eight non-structural proteins. Proteins unique to the Pestivirus genus are the non-structural protease Npro and Erns envelope glycoprotein which has RNase activity. Both proteins are associated with the suppression of the host’s innate antiviral immune response (23). This review compiles current progresses on the roles and functions of Pestivirus Npro in the evasion of type I interferon response.
Npro of pestiviruses has antagonistic activity against type-I IFN productionNpro is the first protein encoded by pestivirus, with a molecular weight of 23 KDa, and is unique to the pestivirus genus. Npro protein is a hydrophilic peripheral membrane protein without signal peptide, and the secondary structure mainly contains β-sheet and random curling. Moreover, it has autoprotease activity and can be cleaved in an autocatalytic manner from nascent polyproteins being translated into mature viral proteins. Npro is not necessary for the replication of pestiviruses but plays an important role in the evasion of the antiviral immune response of host cells. It has been shown that Cys69 and His130 are the catalytic residues of protease cleavage and catalyze the cleavage of peptide bonds between Tyr164 and Vail65. To study the biological activity of Npro, Tratschin et al. prepared a CSFV virus strain vA187-Ubi, the Npro protein gene sequence of which was replaced by the mouse ubiquitin protein gene. It has been shown that vA187-Ubi had similar growth characteristics to the parent vA187-1 virus, both of which showed obvious cytopathological effects. In vivo assay results showed a complete loss of virulence of vA187-Ubi, indicating the Npro protein is unnecessary for the virus replication but is essential for its virulence (25). In addition, the Npro protein of pestiviruses could block apoptosis and IFN-α/β production induced by double-stranded RNA. NCP-type BVDV-1 infection was found to protect bovine nose osteocytes from poly (I: C) -induced apoptosis. Further studies showed that NCP-type BVDV inhibited the transcription and secretion of type I interferon-induced poly (I: C) (26). In addition, compared with the parent strain, ΔNpro BVDV can effectively induce IFN-β production, indicating that Npro could inhibit the production of type I interferon (27). The Npro protein of BVDV-2 could also significantly down-regulate oligo adenylate synthetase (OAS), ubiquitin-like protein 15 (ISG15), Myxoviral-resistant protein 1 (Mx1), and type I IFN transcription levels (28).
Studies on CSFV have shown similar results. Ruggli et al. reported that after infection with CSFV, the resistance to poly (I: C) -induced apoptosis by porcine renal cell line SK-6 increased nearly 100 times. ΔNpro CSFV was found to have a similar growth profile to wild-type virus, but with no protection for SK-6 cells against apoptosis induced by poly (I: C) (29). Therefore, it was suggested that Npro could counteract dsRNA-induced apoptosis and IFN-α/β production independently of other CSFV proteins. After treated with poly (I: C), drastically more SK6-EGFP-Npro cells and CSFV-infected SK-6 cells survived compared with the parental SK-6 cells. Luciferase reporter gene experiments showed that Npro also inhibits the expression of luciferase derived by IFN-α/β promoter in human cells, meanwhile, it can also inhibit the production of Newcastle disease virus-induced IFN-α/β (30). Moreover, in dendritic cells (DC), ΔNpro CSFV can promote the expression of IFNα/β, and also up-regulate the expression of CD80/86 and MHC II to promote the maturation of DCs (31).
Bungowannah virus is genetically the most divergent pestivirus with all of the genomic and structural elements of classical pestiviruses. Compared with other pestivirus, they also have many differences in antigenic cross-reaction. To test the influence of Npro of Bungowannah virus on the type I interferon signaling pathway, a chimeric BVDV/Bungowannah virus (vCP7_Npro-Bungo) was rescued by Richter et al. (32). In the virus, the Npro gene of Bungowannah virus replaced that of CP7— a cytopathic BVDV strain. After infected with CP7, Bungowannah virus, and virus vCP7_Npro-Bungo, similar IFN suppression was observed in cells. However, the Npro-deleted mutant had an impaired replication and induced increased type-I IFN response in bovine cells (32). Collectively, these studies indicated that the Npro of pestiviruses had antagonistic protease activity of IFN-α/β production.
Furthermore, it has been shown that the replacement of amino acids Glu22 and His49 of pestiviruses could abolish the ability of Npro to inhibit IFN production, while the replacement of Cys69 had no such effect. There was no antagonistic IFN-α/β activity in the conserved Npro region (L8P) mutant near the N-terminal of the two BVDV biotypes, demonstrating the integrality of the Npro N-terminal structure is essential in the catalytic activity of IFN-α/β inhibition (33).
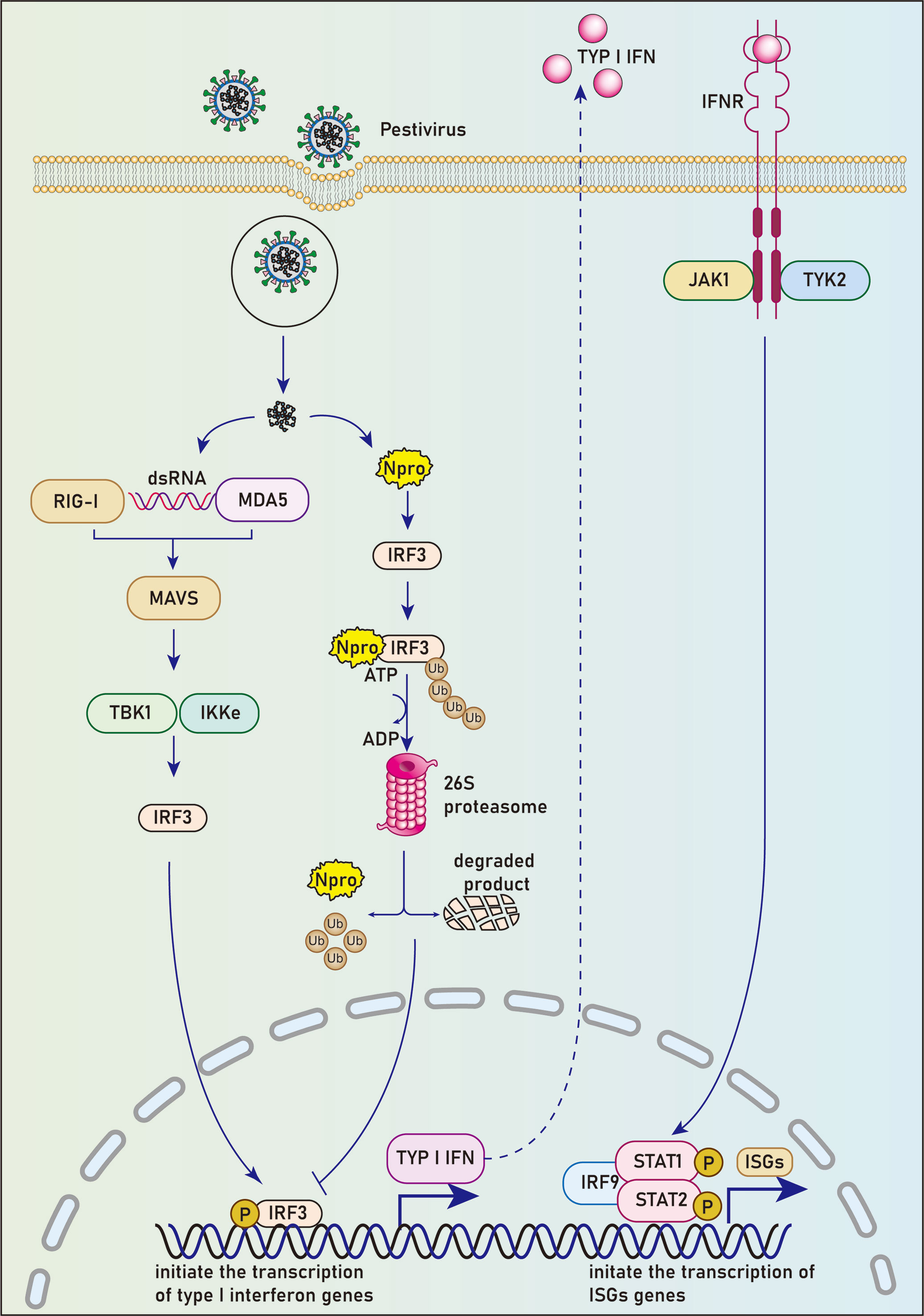
Npro induces proteasome degradation of IRF3Overview of IRF3The IRF family has been reported to have 10 members, namely IRF1-IRF9 and virus IRF (v-IRF). IRF3, a principal transcription factor, is significant in the antiviral immune response (34). IRF3 is highly homologous to IRF7. Both of them regulate the type I IFN synthesis, but play different roles in the innate immune response. IRF3 is critical for early induction of IFN expression in most cells post-viral infection; IRF7, which induces both IFNα and IFNβ expression, has functions in the antiviral activity of IFN in a later stage. In contrast, IRF3 can induce IFNβ gene expression, but not other IFNα expression except IFNα4 (35). Upon viral infection, a series of cellular pathways are activated subsequently to promote the translocation of phosphorylated IRF3 or IRF7 into the nucleus and initiate the transcription of type I interferon genes by attaching to IFN-α/β promoters (36).
Npro mediates ubiquitination and proteasomal degradation of IRF3By luciferase reporter gene experiment, La Rocca et al. found that CSFV-infected cells could inhibit IRF3 gene transcription. The use of cell lines expressing CSFV Npro confirmed that the Npro protein reduced the expression of IRF3, suggesting that this single viral protein specific to the pestiviruses can inhibit interferon production in the innate immune response to the virus (37) (Figure 1). Hilton et al. reported that NCP-BVDV (pe515) infection could induce the translocation of a small amount of IRF-3 from the cytoplasm to the nucleus at the early stage of infection. In addition, most IRF3 in the cytoplasm was degraded by the ubiquitination-proteasome pathway mediated by the Npro protein (27). Similar to the NCP-BVDV virus, CP-BVDV (NADL strain) does not induce interferon response after infection and blocks interferon-stimulating genes induced by paramyxovirus infection, resulting in a significant decrease in IRF3 expression. However, the IRF3 repression activity is considered independent of the protease activity of Npro. Further studies revealed that Npro could interact with IRF3 before its phosphorylation-induced activation, leading to the ubiquitination and proteasomal degradation of IRF3 (38).
Figure 1 Npro blocks the host’s IFN-activated immune response by degradation of IRF3. Upon viral infection, pathogenic associated molecular patterns (PAMPs) are recognized by cellular pattern recognition receptors (PRRs). A series of cellular pathways were activated subsequently to promote the translocation of phosphorylated IRF3 into the nucleus and initiate the transcription of type I interferon genes by binding to IFN-α/β promoters. Npro could interact with IRF3 before it’s phosphorylation-induced activation, leading to the ubiquitination and proteasomal degradation of IRF-3 and subsequent inhibition of the type I interferon response.
The Npro protein of CSFV can also mediate the degradation of IRF3 after interaction with it. However, different from the research results of La Rocca et al., There are other studies reported that CSFV infection does not inhibit a cytomegalovirus (CMV) promoter-driven IRF3 expression. Furthermore, CSFV neither reduces the transcriptional activity of the IRF3 promoter nor affects the stability of IRF3 mRNA (39, 40).
Ubiquitin contains 76 amino acid residues and is highly conserved in all eukaryotes (41). Selective binding of linear ubiquitin to a protein is the initial signal for target protein degradation. Ubiquitin chains can be conjugated to the specific protein substrate through an isopeptide bond between the ubiquitin C-terminal glycine residues and the lysine residues in the substrate. Proteasome-dependent degradation of proteins mediated by ubiquitination regulates a variety of biological reactions in the body, including cell cycle, signaling, DNA repair, and apoptosis (42, 43).
The ubiquitin modification process requires successive events associated with three enzymes: an E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme, and an E3 ubiquitin ligase. Ubiquitin is first connected to the catalytic cysteine residues of endogenous E1 protein by an intermediate thioester bond. It is then conveyed to an E2 conjugating enzyme through a trans-esterification reaction. Subsequently, E3 ubiquitin ligase transfers the activated ubiquitin from an E2 to substrates. Finally, the substrate labeled with ubiquitin is delivered to the 26S proteasome for degradation (44). Once the E1 ubiquitin-activating enzyme was thermal inactivated, the degradation activity of IRF3 mediated by the Npro protein of CP-BVDV was lost (39), indicating that the E1 ubiquitin-activating enzyme is involved in the Npro protein-mediated ubiquitin modification and degradation of IRF3.
Besides contributing to the antiviral immune response, IRF3 participates in the control of the cell cycle, apoptosis, and tumor suppression as well (45). When cells are infected with Sendai virus or stimulated by double-stranded RNA, IRF3 can bind to cytoplasmic Bax via its BH3 region and transfer Bax to mitochondria to activate apoptosis. It has been shown that wild-type Npro could restrain apoptosis signals induced by astrosporin, interferon, double-stranded RNA, sodium arsenate, and hydrogen peroxide, which was mainly achieved by the degradation of IRF3 protein. In addition, the Npro protein can inhibit the Bax-dependent apoptosis pathway by inhibiting mitochondrial lysis and Bax redistribution through interaction with the mitochondrial lumen. Moreover, the Npro protein could rapidly localize to ubiquitin-containing peroxisome. Thus, the Npro protein may bind to IRF3 and degrade it by transporting it to ubiquitin-containing peroxisome (46).
Peptide-prolyl isomerase (Pin1), Ro52 (TRIM21), RBCC protein interacting with PKC1 (RBCK1), RTA-associated ubiquitin ligase (RAUL), Tripartite motif 21 (TRIM21), Forkhead transcription factors of the O class (FOXO1), Casitas B-lineage lymphoma (c-Cbl) have been suggested as E3 ligases to invoke ubiquitination and subsequent degradation of IRF3 in the cytoplasm, thus inhibiting the production of type I IFN (47–51). However, it is unclear which E3 ubiquitin ligase contributes to the ubiquitination degradation of IRF3 mediated by the Npro protein. It is worthy of further exploration whether Npro has crosstalk with these E3 ligases or molecules to regulate the ubiquitination and subsequent degradation of IRF3.
Currently, the Npro binding site on IRF3 is unclear. It has been revealed that Npro can interact with IRF3 directly and forms a soluble 1:1 complex by utilizing recombinant Npro and IRF3 proteins. This interaction requires the complete IRF3 protein rather than any of the single domains, the DBD in N-terminal or the IAD in C-terminal (52). It has been also shown in a previous mammalian two-hybrid analysis that the association of Npro with IRF3 needs both the DBD and the IAD of IRF3 (53). However, the exact arrangement of DBD and the IAD in the IRF3 monomer and dimer is still unknown (35, 54, 55). As the ~60-amino-acid linker region between the two domains is somewhat helical but not structured in the absence of either DBD or IAD (56). The intact linker is thus suggested to be involved in Npro binding (52). Npro has been shown to interact with the IRF3 monomer and phosphomimetic dimer, indicating that the Npro binding site on IRF3 contains areas not affected by the phosphorylation and subsequent activation status of IRF3 (52). Npro can also interact with IRF3 in the complex with its transcriptional cofactor, the CREB-binding protein (CBP). Therefore, the contact surface in the IRF3 dimer and CBP binding site is not required for Npro binding (52).
BVDV Npro protein has been shown to degrade IRF3 in the cytoplasm, whereas IRF3 in the nucleus is resistant to this degradation (26). The influence of cellular localization of Npro on IRF3 degradation is unclear. A recombinant virus vSMS-IRF3 was constructed by inserting the IRF3 gene sequence between the 13th and 14th amino acid sites of the Npro protein of the highly virulent CSFV Shimen strain by Li et, al (57). The fusion protein of IRF3-Npro expressed by the recombinant virus only located in the cytoplasm and vSMS-IRF3 was significantly attenuated. Pigs inoculated with the recombinant virus were all resistant to the lethal CSFV challenge, but the parent virus showed a typical virulent phenotype (57). Therefore, it was suggested that the nuclear localization of Npro is essential to the replication and virulence of CSFV (57).
However, a previous study showed that any mutants of L8P, E22L, and H49V in Npro could abolish its IFN-α/β antagonistic activity, revealing that the 49 amino acids in the N-terminal of Npro protein are necessary to type-I IFN suppression (33). Based on this, we speculate that the insertion of the IRF3 gene into the Npro gene may eliminate its function of IRF3 degradation. Thus, the attenuation of vSM-IRF3 may be caused by the loss of the IFN-α/β antagonistic activity of IRF3-Npro rather than its cytoplasmic localization. Moreover, Npro was observed in the nucleus in a diffuse manner (58, 59), and could bind to IRF3 dimer or the IRF3 dimer in the complex with CBP (52). Therefore, the insertion of IRF3 into Npro could also affect the cellular diffusion of the IRF3-Npro fusion protein, leading to its accumulation in the cytoplasm. The effects of cellular localization of Npro protein on CSFV virulence need further study.
Whether Npro protein is the main determinant of the virulence of pestiviruses is still a controversial topic. Continuous passage of CSFV attenuated vaccine strain GPE- in pigs restored its virulence, but did not regain the ability of its Npro to degrade IRF3 (60). However, strains containing the N136D mutation in Npro restored the IRF3 degradation activity and IFN-α/β antagonistic ability in vitro as well as pathogenicity in vivo. These results demonstrate that the Npro protein makes a decisive contribution to the virulence of pestiviruses, but there are other factors that can regulate the virulence of pestiviruses.
The Npro protein of CSFV can also interact with IRF7 in plasmacytoid dendritic cells, down-regulating the expression level of IRF7 protein and further inhibiting the IFN-α expression. Whereas, the molecular mechanism of Npro inhibiting the expression of IRF7 protein is still unclear. It is certain, however, that this antagonism does not involve either polyubiquitination or protease degradation pathways (53).
The zinc atom binding motif of Npro is critical for the degradation of IRF3Analysis by sequence alignment revealed that the C-terminal half of the Npro protein contains a conserved metal binding TRASH motif composed of Cys-X21-Cys-X3-Cys (where X is any amino acid). TRASH motif commonly exists in proteins associated with heavy metal recognizing, resistance, transcription regulation, cation transportation, and hydrogenase. Inductively coupled plasma–mass spectrometry (ICP-MS) assay indicated that each Npro protein molecule could coordinate a single zinc atom. Site-directed mutagenesis studies revealed that the zinc-binding sites of Npro protein include Cys112, Cys134, Cys138, and probably Asp136. These zinc-binding site mutations lead to the deficit of Npro protein-mediated IRF3 degradation in cells inoculated with CSFV, suggesting that the zinc-binding capacity of Npro protein is critical for virus-mediated IRF3 degradation (61). In addition, the zinc-binding domain of Npro is critical for its protein stability and its interaction with IRF7 (62).
Npro interacts extensively with host proteinsProteins in cytoplasmic ribonucleoprotein particlesRecently, pull-down combined mass spectrometry showed that Npro binds to more than 55 kinds of proteins, mainly RNA helicase A (DHX9), Y-box binding protein (YBX1), DDX3, DDX5, IGF2BP1, eIF3, and other ribosomal proteins, multiple myeloma tumor protein 2, interleukin enhancer binding factor 3 (IEBP3) guanine nucleotide-binding protein 3, and polyadenylate-binding protein 1 (PABP-1). Many of the interacting proteins are components in cytoplasmic ribonucleoprotein particles (RNPs). They play roles in regulating the translation of mRNA and could be recruited into stress granules to regulate the translational initiation rate or mRNA degradation (63). The assembled stress granules might control the proliferation of viruses and some viruses could in turn hinder their formation or even disassemble them (64). It has been suggested that Npro could redistribute to stress granules after interaction with YBX1 through its TRASH domain. When exposed to oxidative stress, cells expressing Npro alone assembled stress granules and Npro colocalized with stress granule proteins. In contrast, the formation of stress granules in NCP-BVDV-infected cells was inhibited, indicating that this inhibition was not caused by Npro binding to ribonucleoproteins (63). Thereby, Npro may not influence the function and location of ribonucleoproteins although it could be localized to stress granules by interacting with these host proteins. As some interacting proteins of Npro are also involved in RISC function during RNA silencing, further studies were conducted to determine whether Npro affected RNA interference (RNAi). However, the outcomes suggested the expression of Npro had no influence on RNAi silencing activity (65).
S100A9S100A9, one of the danger-associated molecular patterns (DAMPs) proteins, is vital in the innate immune system and always accumulates in large amounts in ectocytic space during inflammation responses (66). Additionally, the S100A9 could effectively trigger inflammatory responses through Toll-like receptor 4 (TLR4) as a homodimer (67–73). In the heterodimer with S100A8, the S100A9 exerts antimicrobial activity by inhibiting microbes from acquiring nutrients (74–79). Yet, the function of S100A9 in virus infection is unclear. It has been shown that the amount of S100A9 was increased after treatment with poly (I: C), which is an analog of viral dsRNA (80). The expression of S100A9 is also highly enhanced in human papillomavirus-associated dysplastic tissues (81) and BKV-infected recipients post-kidney transplantation (82). Likewise, high levels of S100A9 expression were observed in lungs and livers in autopsied subjects with COVID-19 and pre-existing chronic liver disease (83). Darweesh et al. reported that NCP-BVDV2a 1373 Npro protein has a strong interaction with cellular S100A9 protein. Furthermore, the Npro protein enhances the replication of BVDV in infected cells by inhibiting S100A9 activity in epidermal cells (84).
TRIM56Currently, the TRIM family consists of more than 60 members and could be divided into 11 subfamilies (85). Although their exact functions are still unclear, the TRIM proteins contribute to a large variety of biological activities, such as cell proliferation, development, differentiation, immunity, apoptosis, and innate immune response to pathogens (86–90). As a protein of the fifth subfamily of TRIM, TRIM56 is expressed in the cytoplasm after type I interferon stimulation (91, 92). A previous study has identified TRIM56 as a cellular protein that associates with BVDV Npro through its C-terminal portion. Although TRIM56 has RING-dependent E3 ubiquitin ligase function, it is not involved in Npro-mediated IRF3 degradation nor degrade Npro. Furthermore, it was suggested that both ectopically and endogenously expressed TRIM56 contribute to impaired replication of BVDV due to its E3 ligase activity. In contrast, the downregulation of TRIM56 expression largely improved BVDV proliferation. Moreover, it is the integrity of the TRIM56 C-terminal, rather than the TRIM56-Npro association that contributes to TRIM56’s antiviral activity (91).
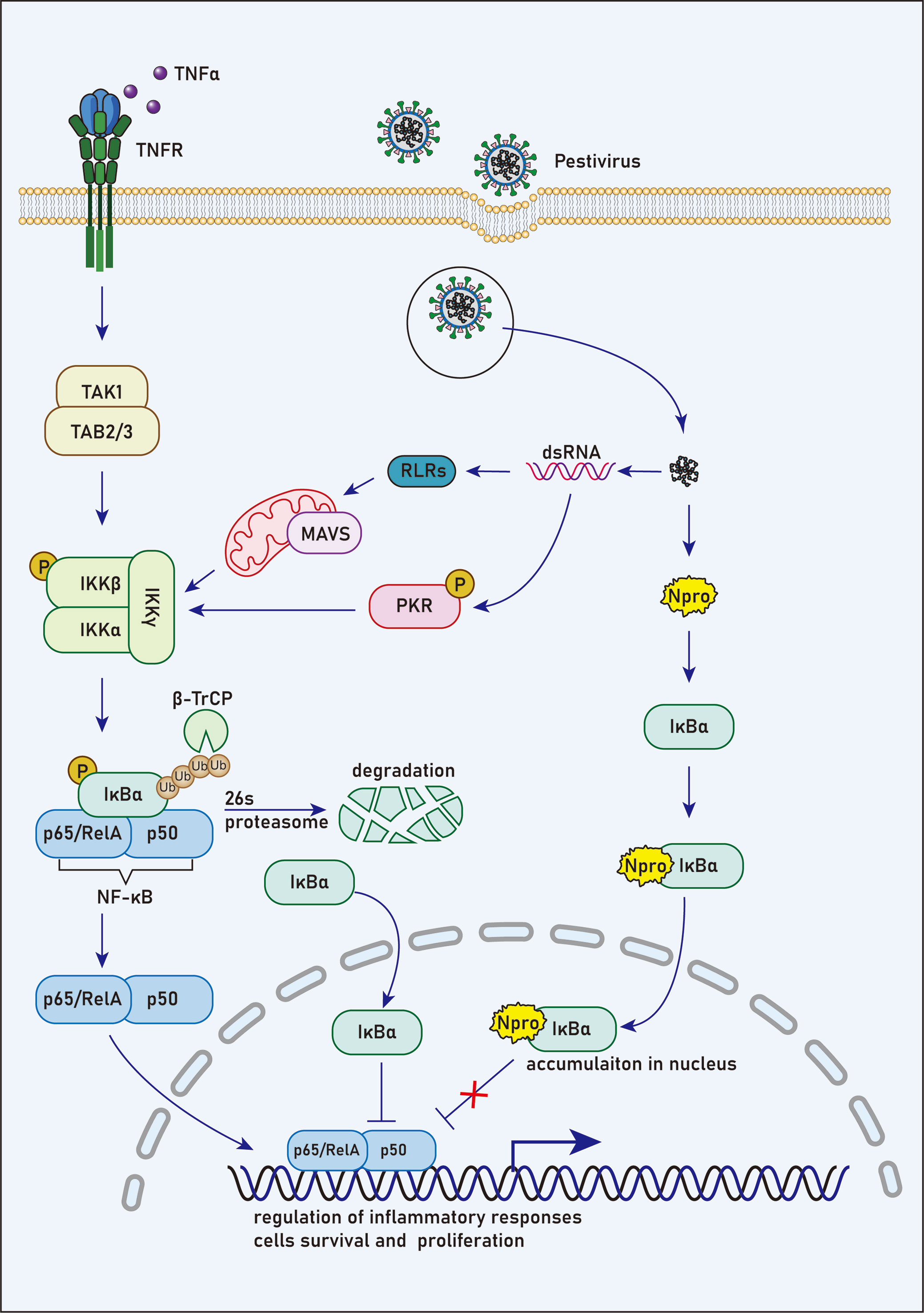
pIκBαTo discover host proteins that could bind to Npro of CSFV, Doceul, et al. (58) conducted a yeast two-hybrid assay of a human library. It was revealed that Npro had a direct association with IκBα, which is responsible for apoptosis regulation, the immune reaction, and IFN expression. As an inhibitor of NF-κB, IκBα is also a prime target for immune evasion strategies developed by many viruses (93–95). Further studies suggested the interaction of Npro with aa 213-317 of the C-terminus of pIκBα (pig IκBα) (58), which also contact with NF-κB through the domain between aa214-280 (96, 97). This suggests that Npro competes with NF-κB for unbound pIκBα (58).
Before stimulation, NF-κB remains in an inactive state in the cytoplasm due to its interaction with IκBα, which covers the nuclear localization signals of NF-κB. It has been reported that NF-κB/IκBα complex could be triggered by phosphorylation upon various stimulation, such as viruses and bacteria (98–101). In this case, IκBα is phosphorylated at Ser32 and Ser36 by the IKKβ subunit following the activation of the IKK complex (IKKα/IKKβ/IKKγ). Then, the E3 ubiquitin ligase complex, SCFβ−TRCP, ubiquitinates IκBα and targets it for degradation by the 26S proteasome, resulting in the release of NF-κB for nuclear translocation (102–105). However, the activated NF-κB initiates regeneration of IκBα, which detaches NF-κB from DNA after its translocation to the nucleus, and conveys NF-κB to the cytoplasm in a nuclear export sequence-dependent process (106–108). Tumor necrosis factor-alpha receptor (TNFR) activated by binding with TNF-α is one of the principal receptors that mediate NF-κB activation (109).
It’s reasonable to speculate that under stimulation of TNF-α, new synthesized cytoplasmic IκBα induced by NF-κB activation could bind to Npro in cells expressing Npro proteins. Therefore, limited unbound IκBα translocate into the nucleus, and thus the suppression of NF-κB DNA-binding activity by IκBα should be restricted (Figure 2). It has been suggested that HIV-1 tat transactivator could activate NF-κB by interacting with IκBα and by inhibiting the repressor from binding to the NF-κB complex (110). However, the ability of the p65 subunit of NF-κB to bind the promoter sequence in CSFV-infected PK15 cells was not affected by functional analysis (111). Furthermore, after TNF-α stimulation of Npro stable expression PK15 cells, a high concentration of pIκBα was observed in the nucleus, but the function and expression of NF-κB did not change significantly (58). Therefore, TNF-α may stimulate the rapid resynthesis and massive nuclear translocation of pIκBα, many of which are bound to Npro and does not affect the action of NF-κB thus resulting in the accumulation of a large amount of pIκBα in the nucleus. However, the effect of Npro binding to pIκBα on the activity of NF-κB in the nucleus and cytoplasm is worth further investigation.
Figure 2 N competes with NF-κB to bind with IκBα. Prior to stimulation, NF-κB remains an inactive state in the cytoplasm due to its interaction with IκBα, which masks the unclear localization signals of NF-κB. NF-κB/IκBα complex is activated by phosphorylation in response to various stimuli, such as viral and bacterial pathogens. In this case, IκBα is phosphorylated at Ser32 and Ser36 by the IKKβ subunit following the activation of IKK complex. Then, the E3 ubiquitin ligase complex, SCFβ−TRCP, ubiquitinates IκBα and targets it for degradation by the 26S proteasome, leading to the release of NF-κB for nuclear translocation. However, NF-κB activation induces rapid resynthesis of IκBα, which translocates to the nucleus, dissociates NF-κB from DNA and transports NF-κB to the cytoplasm in a nuclear export sequence-dependent process.
HAX-1HS-1-associated protein X-1 (HAX-1), a protein of approximately 35-kDa, is universally synthesized in murine and human tissues (112, 113), especially in the mitochondria (114). Primarily, it was shown to play a role in the control of apoptosis or programmed cell death (114). A recent study has indicated that HAX-1 may also contribute to the control of calcium homeostasis and cell survival in cardiac tissue (115). It was found that the protein could bind to the Npro protein of CSFV by yeast two-hybrid, and the interaction between the two proteins was further confirmed by co-immunoprecipitation assay (116). During CSFV infection, the expression level of HAX-1 did not change significantly, indicating that Npro interacting with HAX-1 could not degrade it. However, in the cells co-transfected with HAX-1 and Npro, the two proteins were significantly transported to the endoplasmic reticulum, and the Npro protein in the nucleus was significantly reduced (116). Significantly, the relocation of HAX-1 to the ER in the presence of phospholamban (PLN), a crucial regulator of Ca2+ homeostasis and contractility in the heart, correlated with stronger resistance to apoptosis (117). Therefore, it could be hypothesized that during CSFV infection, Npro protein may bind to HAX-1 in the cytoplasm and transport it to ER to enhance the tolerance of infected cells to apoptosis. Thus, less Npro protein synthesized in the cytoplasm diffuses into the nucleus. However, further research is needed to confirm this hypothesis.
PCBPsAs members of the K homology (KH) domain superfamily, Poly(C)-binding proteins (PCBPs) are known for their interaction with both RNA and DNA specifically. It has been suggested that the KH domain superfamily proteins associate with the stability of cellular mRNAs (118–120), regulate their translation (121–125), and also involve in the host antiviral reaction (126, 127). Among the PCBPs, PCBP1 is an ssDNA-binding protein that contributes to the transcription of the neuronal μ-opioid receptor gene (122). Cytoplasmic and nuclear expression of CBP1 been demonstrated (128). Li et al. found that PCBP1 protein could interact with Npro protein. Knocking down the expression of PCBP1 could inhibit the replication of CSFV, while overexpression of PCBP1 could promote the reproduction of CSFV. PCBP1 inhibits the IFN signaling pathway by degrading MAVS and enhances the replication of CSFV genomic RNA, thus promoting the proliferation of CSFV (129). However, whether the interaction between PCBP1 and Npro has any effect on their cellular localization or CSFV replication needs further exploration.
Response of Npro protein to type-3 interferonNpro has also been suggested to inhibit the innate immune reaction by restricting type III IFNs (IFN-λs). Although many similarities exist between types I and III IFNs in the signaling networks and expression processes (130, 131), a host of distinctions are evident. Different from type I IFNs induction which needs all IFN enhanceosome elements, type III IFNs are induced independent of IRFs or NF-κB (132). Besides, unlike the ubiquitous receptors for type I IFNs, type III IFN receptors are mainly expressed in mucosal epithelia (133). Thus, type III IFNs are considered to be crucial for immune responses in the respiratory and gastrointestinal tracts (134). As IRFs and NF-κB are important regulators for type III IFNs, IRF1 may have a specific function in this process (135). Npro-deficient CSFV has been shown to significantly enhance the transcription level of IFN-λs 24h post-infection. In contrast, overexpression of Npro significantly reduced the IFN-λs transcription and IFN-λ3 promoter activity. Moreover, in poly (I: C)-treated IPEC-J2 cells, overexpression of Npro or infection with wtCSFV not only down-regulated the production and the promoter activity of IRF1 significantly but also inhibited IRF1 nuclear translocation. This suggests that Npro could restrict type III IFNs response by preventing the production and nuclear translocation of IRF1 (134).
ConclusionPestiviruses are counted among the highly destructive and economically important pathogens, which have evolved many strategies to evade elimination by the host antiviral immune response. Studies emphasizing various molecular techniques undertaken during the last two decades have elucidated at least two viral proteins (Npro and Erns RNase) as IFN antagonists of pestiviruses. Unlike repression of the interferon response via the effect of Npro on IRF3, the secreted Erns protein distributed by the bloodstream could be taken up by cells, specifically PDCs, and degrade pestiviral RNA. Therefore, Erns hinders IFN production induced by the extracellular synthetic or viral ss- or dsRNAs (136–138). Thus, we conclude that pestiviruses evade the host’s IFN-activated innate antiviral immune response in a complex way to establish and maintain a persistent infection status. This article mainly reviewed the progress of innate immune evasion mediated by Npro of pestiviruses. while its detailed process concerning blocking the IFN-1 response remains obscure. Further understanding of the approaches employed by viruses of this genus to control immune response to escape the innate immune system is in need, which will eventually contribute to developing effective strategies to prevent and control pestivirus infection.
Author contributionsWriting-original draft preparation: SW and XLi. Writing review: YS, XLv, and KL. Figures: SW. Supervision: JZ. Funding acquisition: SW, YS, JR, and JZ. All authors contributed to the article and approved the submitted version.
FundingThis work was financially supported by the Young Scientific and Technological Talents in Inner Mongolia (No. NJYT23095, NJYT22053). The Natural Science Foundation of Inner Mongolia (No. 2022LHQN03009). Doctoral Funding of Inner Mongolia Minzu University (No. BS584, BS583), Key Research and Development Program in Inner Mongolia Autonomous Region (No. 2021ZD001301, 2019ZD006). Open Funding Project of Brucellosis Prevention and Treatment Engineering Research Center of Inner Mongolia Autonomous Region (No. MDK2021078).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References3. Smith DB, Meyers G, Bukh J, Gould EA, Monath T, Scott Muerhoff A, et al. Proposed revision to the taxonomy of the genus pestivirus, family flaviviridae. J Gen Virol (2017) 98:2106–12. doi: 10.1099/jgv.0.000873
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Postel A, Hansmann F, Baechlein C, Fischer N, Alawi M, Grundhoff A, et al. Presence of atypical porcine pestivirus (APPV) genomes in newborn piglets correlates with congenital tremor. Sci Rep (2016) 6:27735. doi: 10.1038/srep27735
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Postel A, Schmeiser S, Oguzoglu TC, Indenbirken D, Alawi M, Fischer N, et al. Close relationship of ruminant pestiviruses and classical swine fever virus. Emerg Infect Dis (2015) 21:668–72. doi: 10.3201/eid2104.141441
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hause BM, Collin EA, Peddireddi L, Yuan F, Chen Z, Hesse RA, et al. Discovery of a novel putative atypical porcine pestivirus in pigs in the USA. J Gen Virol (2015) 96:2994–8. doi: 10.1099/jgv.0.000251
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kirkland PD, Frost MJ, Finlaison DS, King KR, Ridpath JF, Gu X. Identification of a novel virus in pigs–bungowannah virus: A possible new species of pestivirus. Virus Res (2007) 129:26–34. doi: 10.1016/j.virusres.2007.05.002
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Liu L, Kampa J, Belák S, Baule C. Virus recovery and full-length sequence analysis of atypical bovine pestivirus Th/04_KhonKaen. Vet Microbiol (2009) 138:62–8. doi: 10.1016/j.vetmic.2009.03.006
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Schirrmeier H, Strebelow G, Depner K, Hoffmann B, Beer M. Genetic and antigenic characterization of an atypical pestivirus isolate, a putative member of a novel pestivirus species. J Gen Virol (2004) 85:3647–52. doi: 10.1099/vir.0.80238-0
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Shi K, Xie S, Sun W, Liu H, Zhao J, Yin Y, et al. Evolution and genetic diversity of atypical porcine pestivirus (APPV) from piglets with congenital tremor in guangxi province, southern China. Vet Med Sci (2021) 7:714–23. doi: 10.1002/vms3.407
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Şevik M. Genomic characterization of pestiviruses isolated from bovine, ovine and caprine foetuses in Turkey: A potentially new genotype of pestivirus I species. Transbound Emerg Dis (2021) 68:417–26. doi: 10.1111/tbed.13691
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Chen M, Liu M, Liu S, Shang Y. HoBi-like pestivirus infection leads to bovine death and severe respiratory disease in China. Transbound Emerg Dis (2021) 68:1069–74. doi: 10.1111/tbed.13832
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Pan S, Yan Y, Shi K, Wang M, Mou C, Chen Z. Molecular characterization of two novel atypical porcine pestivirus (APPV) strains from piglets with congenital tremor in China. Transbound Emerg Dis (2019) 66:35–42. doi: 10.1111/tbed.13029
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Becher P, Orlich M, Shannon AD, Horner G, König M, Thiel HJ. Phylogenetic analysis of pestiviruses from domestic and wild ruminants. J Gen Virol (1997) 78(Pt 6):1357–66. doi: 10.1099/0022-1317-78-6-1357
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Vilcek S, Ridpath JF, Van Campen H, Cavender JL, Warg J. Characterization of a novel pestivirus originating from a pronghorn antelope. Virus Res (2005) 108:187–93. doi: 10.1016/j.virusres.2004.09.010
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Neill JD, Ridpath JF, Fischer N, Grundhoff A, Postel A, Becher P. Complete genome sequence of pronghorn virus, a pestivirus. Genome Announc (2014) 2(3):e00575-14. doi: 10.1128/genomeA.00575-14
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Tratschin JD, Moser C, Ruggli N, Hofmann MA. Classical swine fever virus leader proteinase npro is not required for viral replication in cell culture. J Virol (1998) 72:7681–4. doi: 10.1128/jvi.72.9.7681-7684.1998
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Schweizer M, Peterhans E. Noncytopathic bovine viral diarrhea virus inhibits double-stranded RNA-induced apoptosis and interferon synthesis. J Virol (2001) 75:4692–8. doi: 10.1128/jvi.75.10.4692-4698.2001
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Hilton L, Moganeradj K, Zhang G, Chen YH, Randall RE, McCauley JW, et al. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J Virol (2006) 80:11723–32. doi: 10.1128/jvi.01145-06
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Tao J, Liao J, Wang J, Zhang X, Zhang Q, Zhu L, et al. Pig BVDV-2 non-structural protein (N(pro)) links to cellular antiviral response in vitro. Virus Genes (2017) 53:233–9. doi: 10.1007/s11262-016-1410-2
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Ruggli N, Tratschin JD, Schweizer M, McCullough KC, Hofmann MA, Summerfield A. Classical swine fever virus interferes with cellular antiviral defense: Evidence for a novel function of n(pro). J Virol (2003) 77:7645–54. doi: 10.1128/jvi.77.13.7645-7654.2003
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Ruggli N, Bird BH, Liu L, Bauhofer O, Tratschin JD, Hofmann MA. N(pro) of classical swine fever virus is an antagonist of double-stranded RNA-mediated apoptosis and IFN-alpha/beta induction. Virology (2005) 340:265–76. doi: 10.1016/j.virol.2005.06.033
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Bauhofer O, Summerfield A, McCullough KC, Ruggli N. Role of double-stranded RNA and npro of classical swine fever virus in the activation of monocyte-derived dendritic cells. Virology (2005) 343:93–105. doi: 10.1016/j.virol.2005.08.016
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Mou C, Pan S, Wu H, Chen Z. Disruption of interferon-β production by the n(pro) of atypical porcine pestivirus. Virulence (2021) 12:654–65. doi: 10.1080/21505594.2021.1880773
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Richter M, König P, Reimann I, Beer M. N pro of bungowannah virus exhibits the same antagonistic function in the IFN induction pathway than that of other classical pestiviruses. Vet Microbiol (2014) 168:340–7. doi: 10.1016/j.vetmic.2013.11.038
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Gil LH, Ansari IH, Vassilev V, Liang D, Lai VC, Zhong W, et al. The amino-terminal domain of bovine viral diarrhea virus npro protein is necessary for alpha/beta interferon antagonism. J Virol (2006) 80:900–11. doi: 10.1128/jvi.80.2.900-911.2006
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol (2003) 10:913–21. doi: 10.1038/nsb1002
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Honda K, Taniguchi T. IRFs: Master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol (2006) 6:644–58. doi: 10.1038/nri1900
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Baigent SJ, Goodbourn S, McCauley JW. Differential activation of interferon regulatory factors-3 and -7 by non-cytopathogenic and cytopathogenic bovine viral diarrhoea virus. Vet Immunol Immunopathol (2004) 100:135–44. doi: 10.1016/j.vetimm.2004.04.003
PubMed Abstract | CrossRef Full Text | Google Scholar
38. La Rocca SA, Herbert RJ, Crooke H, Drew TW, Wileman TE, Powell PP. Loss of
留言 (0)