
記住我
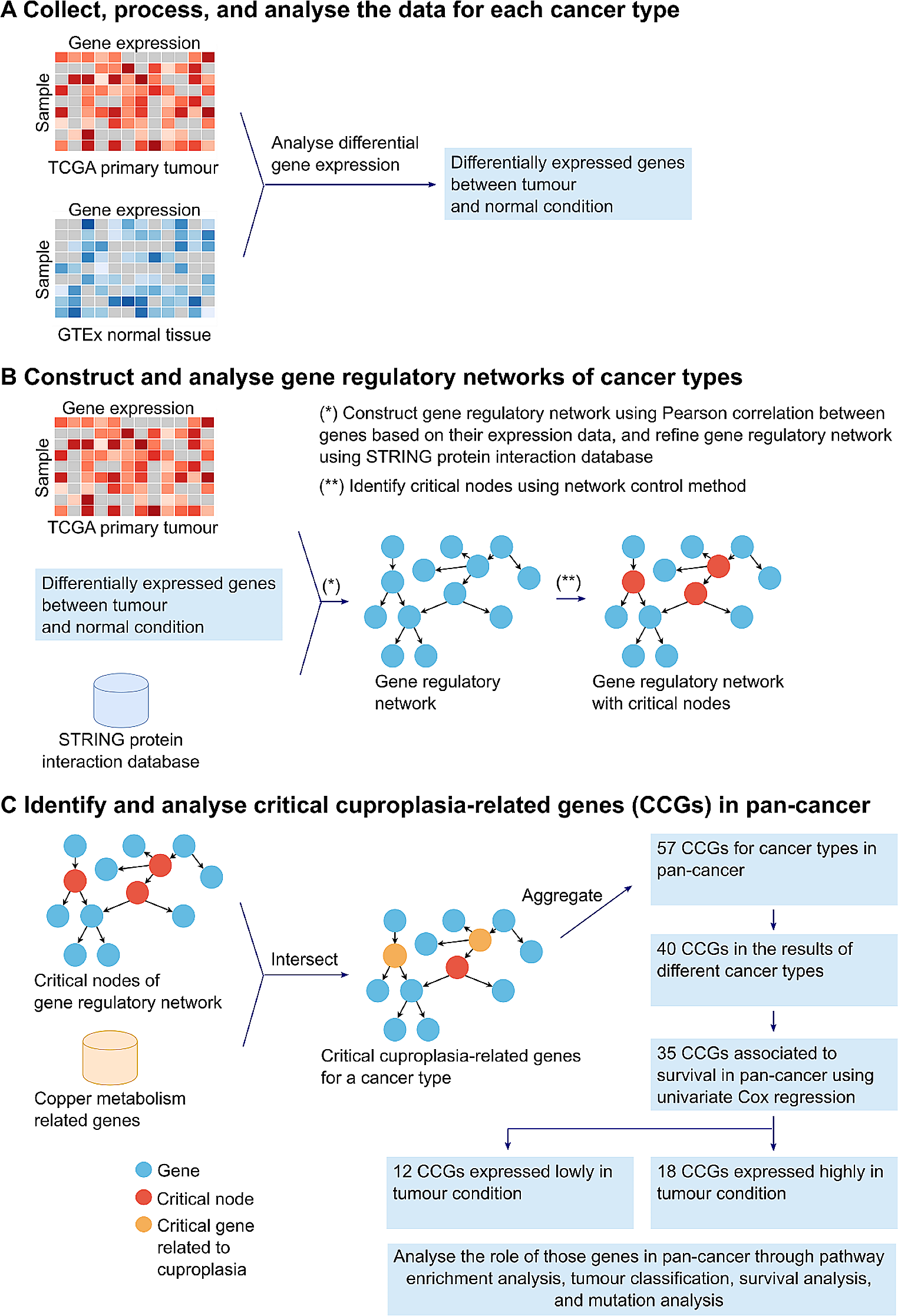


Family 1 is a consanguineous family with the 12-, 6- and 5-year-old brothers III.1, III.2 and III.3 referred for medical evaluation due to multiple congenital joint contractures (Fig. 2a, Table 1). Pregnancy history and birth parameters were not available. Individual III.1 showed movement restriction of shoulder, knee and ankle joints as well as limitations in elbow flexion, internal hip and head rotation. Additional clinical features included microcephaly, camptodactyly, scapular winging, scoliosis, pectus excavatum, reduced palmar and plantar skin folds and translucent skin. Due to finger contractures, surgery was performed. Motor development was generally normal, but reduced fine motor skills and poor active speech were observed. Spinal MRI revealed a cleft formation in the anterior atlas arch. Facial dysmorphism included long face, downslanted palpebral fissures, bifid uvula, high narrow palate, micrognathia and webbed neck. At the last follow-up at the age of 12 years, his weight was 35.5 kg (− 1.0 SD), his length was 145 cm (− 1.1 SD) and his head circumference was 51.8 cm (− 2.1 SD). Physical examination of individual III.2 revealed limitations in shoulder, elbow extension, hip, knee and head rotation, camptodactyly and shortening of the entire dorsal leg muscles with a left club foot and an equinus position of the right foot. Club foot was operated on at the age of 5 years. Similarly to his affected brother III.1, he presented with long face, downslanted palpebral fissures, bifid uvula, high narrow palate, micrognathia, webbed neck, scapular winging, scoliosis, reduced palmar and plantar skin folds and translucent skin (Fig. 1a). Motor and speech development were mildly delayed. Growth measurements at the age of 6 years revealed a body weight of 22.0 kg (− 0.1 SD), a body length of 127 cm (1.4 SD) and a head circumference of 51.0 cm (− 1.5 SD). Individual III.3 showed a less severe phenotype than his brothers. He also presented with movement restrictions mainly in shoulder and elbow, whereas hip and knee are mildly affected. Additional clinical characteristics included scoliosis, camptodactyly, reduced palmar and plantar skin folds, translucent skin as well as congenital renal hypoplasia of the left side. Motor and speech development were mildly delayed. Craniofacial features were similar to those of his brothers with long face, downslanted palpebral fissures, mild micrognathia and webbed neck. Cardiac investigations in all affected brothers revealed no anomalies.
Fig. 2
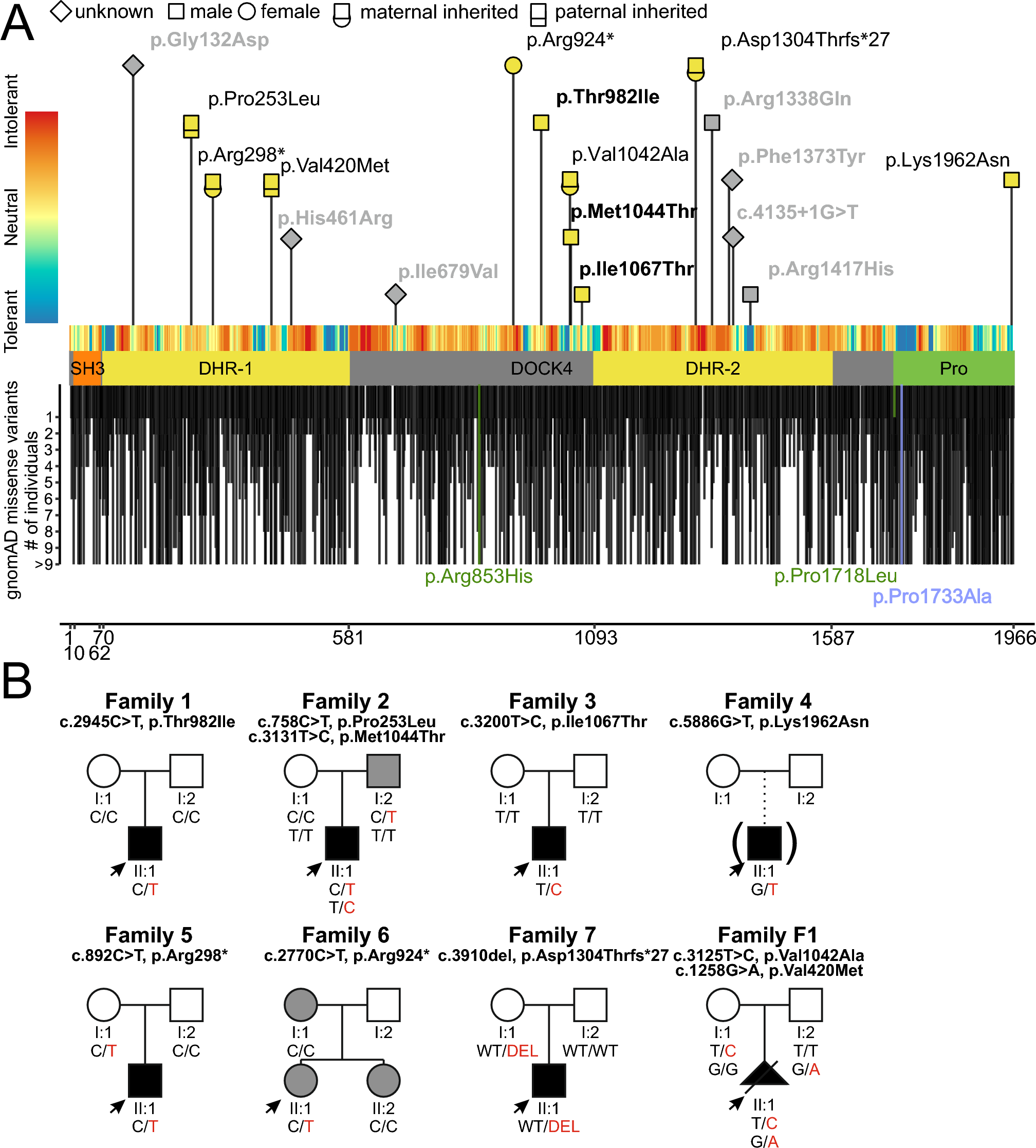
Pedigrees and genetic characteristics of individuals with congenital arthrogryposis and microcephaly carrying homozygous disease-causing variants in FILIP1. a Pedigrees of families 1–3 with pathogenic FILIP1 variants. Affected siblings (solid symbols) in each family carry homozygous disease-causing variants in FILIP1 while non-affected parents and siblings (semi-solid symbols) are heterozygous for identified FILIP1 variants. b Chromatograms of the identified FILIP1 variants in family 1 (F1: c.463G>T; p.Glu155*) and family 2 (F2: c.2665C>T; p.Arg889*) showing homozygosity in affected patients (Mut/Mut) and heterozygous carrier state in healthy parents or siblings (WT/Mut). Localization of nonsense variants is indicated in red. c Copy number analysis by qPCR was used for segregation analysis of a ~ 86-kb homozygous deletion spanning FILIP1 exons 3–6 in family 3 (F3: deletion of Ex3–6) which was initially detected in exome sequencing data of the patient. qPCR for exons 4 and 5 confirmed homozygous deletion of this region in the patient and a heterozygous carrier state in his parents compared to exon 2, which was used as reference
Table 1 Clinical features of patients with disease-causing variants in FILIP1Family 2 is a consanguineous family from Oman. Individual III.3 is a 7-year-old male and was born as the third child to double cousin parents (Fig. 2a, Table 1). During pregnancy, the mother noted decreased fetal movements, otherwise pregnancy and delivery were unremarkable. His birth weight was 2.9 kg (− 1.3 SD), his birth length 56 cm (1.8 SD), and his head circumference 33 cm (− 1.8 SD). Arthrogryposis affecting elbow, wrist, fingers and feet was noted on both sides. In addition, he presented with muscular hypotonia, kyphoscoliosis, reduced palmar and plantar skin folds as well as club feet. Facial dysmorphism included facial hypotonia, triangular facies, low anterior hairline, downslanted palpebral fissures and micrognathia (Fig. 1b). During early childhood, he did not have any major developmental delay apart from difficulties in using his hands. He started sitting without support at the age of 10 months, walking without support at the age of 16 months and spoke his first words at the age of 12 months. Stanford Binet IQ assessment (Janzen et al. 2004) at the age of 5 years showed normal mental function. Electrocardiogram revealed a normal sinus rhythm with normal interval and repolarization, while echocardiography showed normal ejection fraction and mild left ventricular dilation. Investigations of the patient including routine blood investigations, CK levels, liver function tests, brain imaging, and chromosomal microarray were within normal limits. Currently, his weight is 19.9 kg (− 1.7 SD), his length is 123 cm (− 0.6 SD) and his head circumference is 48.5 cm (− 3.2 SD).
Family 3 is of Indian origin and the parents are consanguineous. Individual III.3 is the third child of healthy parents (Fig. 2a, Table 1). The mother’s previous pregnancy ended in intrauterine fetal death and oligohydramnios. After an uneventful pregnancy, the patient was born at term via lower segment cesarean section. He had a birth weight of 2.3 kg (− 2.3 SD). On day 3 of life, he was noticed to have icterus with pale stools, dark yellow colored urine, and distended gall bladder. On clinical examination at day 28 of life, his body weight was 3.5 kg (− 1.2 SD), his length was 48 cm (− 3.4 SD), his head circumference was 36 cm (− 1.2 SD). He was evaluated again at 11 months of life when he had a weight of 6.7 kg (− 3.5 SD), a length of 66 cm (− 3.2 SD) and head circumference of 42.5 cm (− 4.0 SD). He attained neck holding by 3 months of age, sitting without support by 9 months of age, and walking without support by age 1 year 6 months with ability to climb five to six steps. He was able to speak monosyllables by 9 months and bisyllables by 10 months. At 1 year and 10 months of age, it was noted that he can speak a few words but cannot form full sentences. Moreover, he presented with microcephaly, limited movement of wrist and elbow joints, and rocker bottom feet due to which he has an impaired gait (Fig. 1c). Facial features involve epicanthal folds, prominent nose with a broad tip, anteverted nares, long and broad philtrum, short columella, small mouth, and short and broad neck. In addition, prominent veins over abdomen and cholestatic jaundice with scleral icterus were noted. Biochemical blood investigations revealed increased levels of total and direct bilirubin, serum aspartate transaminase, serum alanine transaminase and alkaline phosphatase as well as macrocytic anemia and neutrophilic leukocytosis. Cardiac investigation including electrocardiogram and echocardiography was normal.
Genetic resultsIn family 1, based on the parental consanguinity, we focused on homozygous variants and filtered the ES data of the two affected brothers III.1 and III.2 for variants with coverage of more than 6 reads, a minimum quality score of 10, an allele frequency ≥ 75%, a minor allele frequency (MAF) < 1.0% in the gnomAD database (http://gnomad.broadinstitute.org/), and not annotated in the in-house WES datasets of the CCG. Using these filter criteria, we identified only one homozygous truncating variant shared by the bothers III.1 and III.2 that is predicted to have a severe impact on protein function and to be most likely deleterious. The variant, c.463G>T, is located in exon 4 of the FILIP1 gene (GenBank: NM_015687.4) and induces the formation of a premature stop codon at the amino acid position 155 (p.(Glu155*)) (Fig. 3). The variant is absent from gnomAD database and is classified as pathogenic according to ACMG guidelines (Richards et al. 2015; criteria PVS1, PM2_sup, PM3_sup). In line with the reported parental consanguinity, FILIP1 is embedded within a homozygous stretch of 56.5 Mb (located between positions chr6:36,765,355 and chr6:93,257,601) in individual III.1 and a homozygous stretch of 29.2 Mb (located between positions chr6:49,969,544 and chr6:79,202,472) in individual III.2. We confirmed the presence of the homozygous variant in all three affected brothers III.1, III.2 and III.3 as well as the heterozygous carrier status of the parents by Sanger sequencing (Fig. 2b).
Fig. 3
Schematic representation of the genomic (upper panel) and protein structure (lower panel) of FILIP1. Introns are shown by black horizontal line, coding exons by black bars, non-coding regions of exons by grey bars (upper panel). FILIP1 protein contains a cortactin-binding protein-2 domain (CortBP2) and a central structural maintenance of chromosome (SMC) domain as well as FLNa- and RhoD-binding motifs (lower panel). Localization of the identified nonsense variants in families 1 and 2 (F1; F2) and the deletion of exons 3–6 in family 3 (F3) is marked in red on genomic (RefSeq NM_015687.4) and protein level (RefSeq NP_056502.1)
In family 2, after exclusion of pathogenic or likely pathogenic variants in any of the known genes associated with Mendelian-inherited disorders, ES data of patient III.3 was subsequently analyzed for novel putative causative variants. This analysis revealed the homozygous variant c.2665C>T in FILIP1 that induces the formation of a premature stop codon at the amino acid position 889 (p.(Arg889*)) in exon 5 (Fig. 3). The variant was described in 3/251412 alleles (MAF = 0.00001193) in the gnomAD database, but only present in heterozygous state. Based on ACMG guidelines (Richards et al. 2015; criteria PVS1, PM2_sup, PM3_sup), the variant was classified as pathogenic. We confirmed the homozygous variant in individual III.3 by Sanger sequencing, and co-segregation analysis revealed heterozygous carrier status of both parents and all healthy siblings (Fig. 2b).
In family 3, clinically significant single nucleotide variations or small insertion-deletions correlating with the observed phenotype were not detected in exome sequencing analysis of patient III.3. Interestingly, CNV analysis revealed a ~ 86-kb homozygous deletion (chr6:75,288,632–75,374,566) spanning FILIP1 exons 3–6 (Fig. 3). The deletion is not annotated in the Decipher database (https://decipher.sanger.ac.uk/). In accordance with the ACMG guidelines (Richards et al. 2015; criteria PVS1, PM2_sup, PM3_sup), the deletion was classified as pathogenic. Quantitative PCR showed no amplification of FILIP1 exons 4 and 5 in the patient confirming the homozygous pattern of the deletion, while amplification values in the parents corresponded to one copy number in a heterozygous deletion. No significant differences in amplification values were detected in FILIP1 exon 2 between the patient, his parents and the unrelated control (Fig. 2c).
留言 (0)