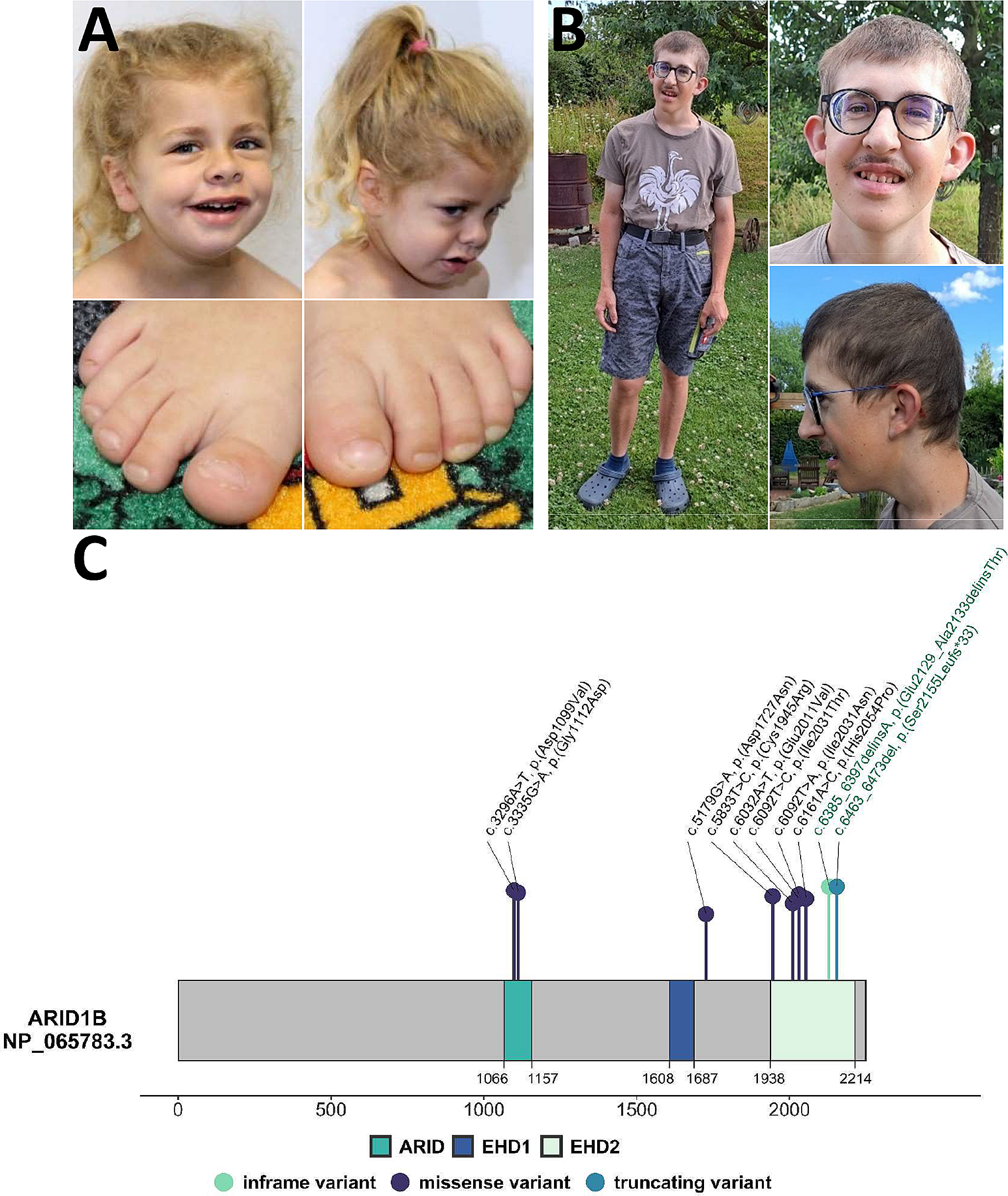
Affected individual
Peripheral blood samples were taken from the affected individual and his parents. In addition, a skin biopsy was obtained from the affected individual and dermal fibroblasts were cultivated according to standard procedures.
Genome sequencing
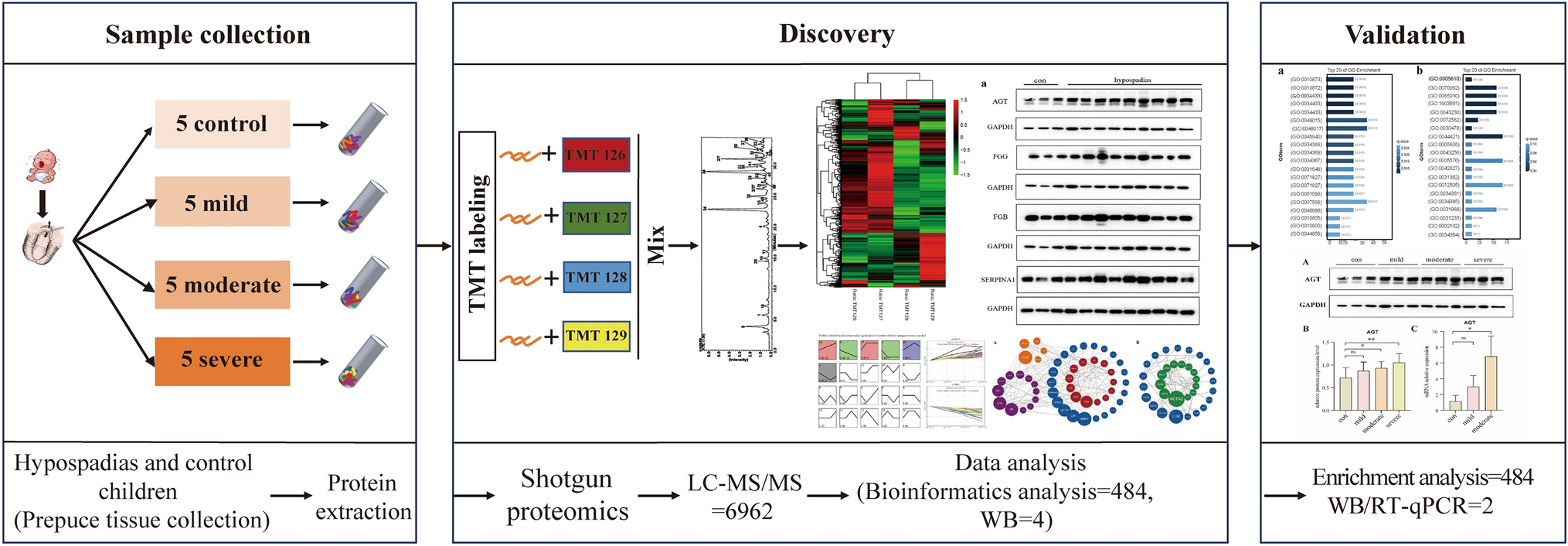
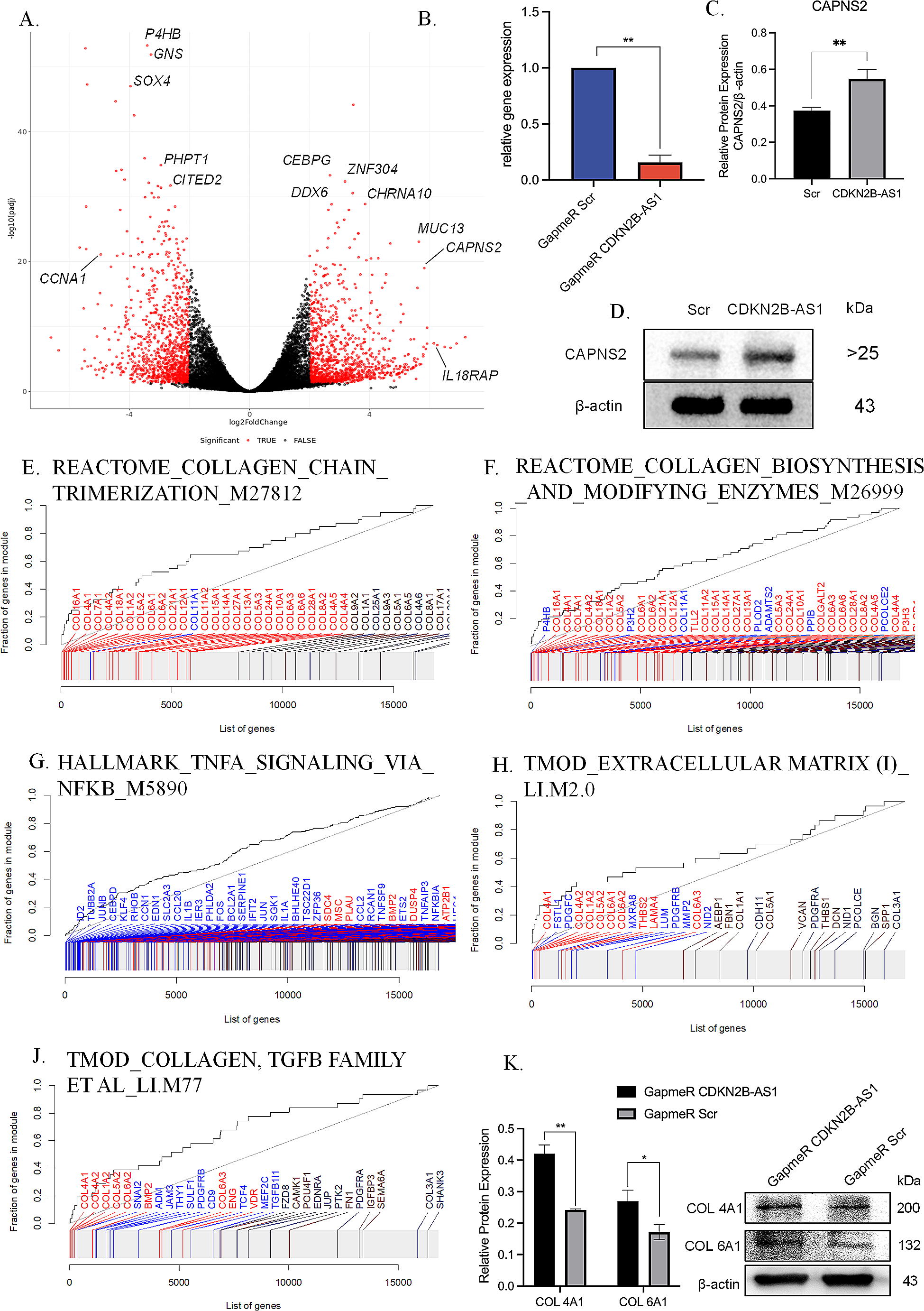
Genome sequencing was performed on the DNA samples from individuals I-1, I-2 and II-1. Libraries were prepared with the DNA tagmentation based library preparation kit (Illumina) without PCR, with 500 ng gDNA input. Library preparation was followed by clean up and/or size selection using SPRI beads (Beckman Coulter Genomics). After library quantification (Qubit, Life Technologies) and validation (Agilent Tape Station), equimolar amounts of library were pooled. The library pools were quantified using the Peqlab KAPA Library Quantification Kit and the Applied Biosystems 7900HT Sequence Detection System and then sequenced on an Illumina NovaSeq6000 sequencing instrument with a paired-end 2 × 150 bp protocol (Target coverage 300x or 1200 Gb per sample). Sequence reads were mapped to the genome version GRCh37 (UCSC hg19) with the Burrows-Wheeler Aligner (BWA MEM). Single-nucleotide variants and short indels were called with the Genome Analysis Toolkit (GATK) according to the GATK Best Practices (McKenna et al. 2010; DePristo et al. 2011). We used Jannovar (Jäger et al. 2014) for variant annotation, and Varfish for filtering and further data analysis as described previously (Holtgrewe et al. 2020).
Sanger sequencing
Sanger sequencing was performed to validate the variants in SUPT7L in DNA samples from the proband and his parents. Exon 2 and 3 of SUPT7L were amplified using the FIREPol Mastermix (SOLIS BIODYNE, Tartu, Estonia) in a ProFlex PCR System (Thermo Fisher Scientific, Dreieich, Germany). Sequencing was performed using BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Dreieich, Germany) and electrophoresis was carried out on an ABI 3730 DNA Analyzer (Thermo Fisher Scientific, Dreieich, Germany). All primer sequences are listed in Supplementary Table 1.
Cell culture
Dermal fibroblasts and HeLa cell line were cultured in DMEM (4.5 g/l glucose, Gibco, Thermo Fisher Scientific, Dreieich, Germany) with 10% fetal calf serum (Gibco, Thermo Fisher Scientific, Dreieich, Germany), 1% UltraGlutamine (Lonza, Basel, Switzerland) and 1% penicillin/streptomycin (Lonza, Basel, Switzerland) at 37°C and 5% CO2.
RNA extraction and cDNA synthesis
Cells were lysed in Trizol (Thermo Fisher Scientific, Dreieich, Germany) and total RNA was extracted using the Direct-Zol RNA Miniprep kit (Zymo Research, Freiburg, Germany). cDNA was transcripted using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Dreieich, Germany).
Qualitative RT-PCR
cDNAs were amplified with primers in exon 2 and 4 of SUPT7L using the FIREPol Mastermix (SOLIS BIODYNE, Tartu, Estonia) in a ProFlex PCR System (Thermo Fisher Scientific, Dreieich, Germany). After Agarose-Gel-Electrophoresis a gel extraction of the amplified fragments S1 of II-1 was performed using the QIAquick Gel Extraction Kit (Qiagen, Venlo, Netherlands). Sequencing of these products were performed as described above. All primer sequences are listed in Supplementary Table 1.
Quantitative RT-PCR
Quantitative PCR was performed on cDNA samples (generated as described above) and carried out using Eva Green (Solis BioDyne, Tartu, Estonia) on a QuantStudio 03 system (Thermo Fisher Scientific, Dreieich, Germany) using two different primer pairs. All primer sequences are listed in Supplementary Table 1.
RNA sequencing and bioinformatics
We performed a poly-A (pA) enrichment from total RNA preparations (II-1 (three technical replicates) and three unaffected controls). Libraries were prepared using the Illumina Stranded TruSeq RNA sample preparation protocol. Library preparation started with 500 ng total RNA. After poly-A selection (using poly-T oligo-attached magnetic beads), mRNA was purified and fragmented using divalent cations under elevated temperature. The RNA fragments underwent reverse transcription using random primers. This was followed by second strand cDNA synthesis with DNA Polymerase I and RNase H. After end repair and A-tailing, indexing adapters were ligated. The products were then purified and amplified (15 PCR cycles) to create the final cDNA libraries. After library validation and quantification (Agilent Tape Station), equimolar amounts of library were pooled. The pools were quantified by using the Peqlab KAPA Library Quantification Kit and the Applied Biosystems 7900HT Sequence Detection System. The pools were sequenced on an Illumina NovaSeq6000 sequencing instrument (Illumina, SanDiego, CA, USA) with a PE100 protocol. 60–70 million sequence reads were generated. RNA-Seq reads were mapped to the human genome (GRCh38.p7) with STAR version-2.7.9a (Dobin et al. 2013). Reads were assigned to genes using FeatureCounts SUBREAD version-v2.0.1 (Liao et al. 2014). For the differential expression analyses we used DESeq2 version-1.34.0 (Love et al. 2014). The gene set enrichment analysis was carried out using CERNO algorithm from R tmod package version-0.46.2 (Zyla et al. 2019). All results are listed in Supplementary Table 2.
Immunoblot
Proteins were extracted in RIPA buffer (150 mM NaCl, 50 mM Tris, 5 mM EDTA, 1% Triton X-100, 0.25% Desoxycholate, 5% SDS) containing protease inhibitor (cOmplete, Roche, Basel, Switzerland). 20 µg of protein per lane was separated by SDS-PAGE, transferred to nitrocellulose membrane and probed with primary antibodies. Immunoblot staining was performed for SUPT7L (rabbit anti-SUPT7L; 25606-1-AP, Proteintech, Rosemont, Illinois, USA), SUPT3H (mouse anti-SPT3, #sc-101157, Santa Cruz Biotechnology, Dallas, Texas), TAF10 (rabbit anti-TAF10, ab263967, Abcam, Cambridge, UK), Lamin A/C (mouse anti-LAMIN A/C; NB100-74451, Novusbio, Centennial, Colorado, USA) and GAPDH (anti-GAPDH, #AM4300, ThermoFisher, Massachusetts, USA). Membranes were incubated with IRDye-/ HRP-conjugated secondary antibodies. Signals were detected with OdysseyFc Imaging System and densitometric quantification was performed using Image Studio (LI-COR Biosciences, Lincoln, Nebraska USA).
Immunofluorescence
Dermal fibroblasts were grown on glass coverslips overnight. Fixation was performed for 10 min in 4% paraformaldehyde at room temperature. The cells were permeabilized using 0.4% Triton X-100 in 3% BSA in 1x PBS for 10 min at room temperature. Immunofluorescence staining was performed for SUPT7L (rabbit anti-SUPT7L; 25606-1-AP, Proteintech, Rosemont, Illinois, USA), LAMIN A/C (mouse anti-LAMIN A/C; NB100-74451, Novusbio, Centennial, Colorado, USA), γH2A.X (mouse anti-pH2A.X (Ser139)(#05-636-I, Merck, Darmstadt, Germany) and turbo-GFP (rabbit anti-turboGFP (#TA150071 Origene, Rockville, Maryland, USA) overnight in 3% BSA in 1x PBS. Secondary antibody staining was performed using anti-mouse IgG Alexa Fluor 488 (#A21202, Invitrogen Waltham, Massachusetts USA) and anti-rabbit IgG Alexa Fluor 555 (#A21572, Invitrogen, Waltham, Massachusetts USA) for 1 h in 1x PBS at room temperature. DNA was stained by DAPI and cells were mounted in Fluoromount G (Biozol, Eching, Germany). Pictures were taken using a LSM700 (Zeiss, Oberkochen, Germany). Each experiment was performed three times.
Cloning of CRISPR/Cas9 plasmid and generation of SUPT7L knockout HeLa cells
The single guide RNA (sgRNA) targeting the third exon of human SUPT7L was designed using the Benchling sgRNA design tool (www.benchling.com) and was selected based on its predicted on- and off-target scores. The sgRNA was then cloned into the PX459 vector from Addgene (#62,988) as previously described (Ran et al. 2013). The final vector was transfected using jetPEI (Polyplus, Illkirch, France) into HeLa EM2-11th cells (Weidenfeld et al. 2009) and 48 h post transfection positive clone selection was initiated with 1.5 µg/ml Puromycin (Thermo Fisher Scientific, Dreieich, Germany). Puromycin-selected single cell clones were expanded and validated by Sanger sequencing. One cell clone carrying a single nucleotide deletion 4 base pairs upstream of the PAM sequence (c.226delC) was selected and further characterized. The deletion causes a frameshift and premature stop codon leading to the gene knockout (KO) (Supplementary Fig. 1).
Cell proliferation assay
Cell proliferation was measured using a WST-1 proliferation assay (Roche, Basel, Switzerland) according to manufacturer’s instructions. In brief, sextuplicates of each cell line (1 × 104 cells/well) were seeded in microtiter plates in 100 µl medium in a humidified atmosphere (37 °C, 5% CO2) for 24 h. Then, 10 µl/well Cell Proliferation Reagent WST-1 were added and incubated for another 2 h. Before measurement, plates were shaken for 1 min and OD/absorbance was determined at 450 nm with 690 nm as reference against blank as background control on a microtiter plate reader (Infinite 200 pro, Tecan).
Analysis of DNA damage
For analyzing DNA damage in dermal fibroblasts (II-1 and unaffected controls) and HeLa cells (WT and SUPT7L-KO), cells were seeded on glass coverslips and handled as described above. γH2A.X (Ser139), a marker for DNA damage (Paull et al. 2000), was stained in the cells and the number of cells with more than six foci of γH2A.X in the nucleus was quantified using immunofluorescence analyses. At least 100 cells per sample were counted and the experiment was performed three times.
Rescue of the DNA damage phenotype
Dermal fibroblasts from the affected individual, unaffected controls and HeLa cells (WT and SUPT7L-KO) were transfected with 2 µg of pCMV6-AC-GFP + SUPT7L-WT (NM_014860.3, Origene) using the Amaxa Nucleofector® 2b (Lonza, Basel, Switzerland) according to manufacturer’s instructions. Immunofluorescence staining of turboGFP and γH2X.A was performed as described above. At least 100 transfected and untransfected cells per sample were counted and the experiment was performed three times.
Statistical analysis
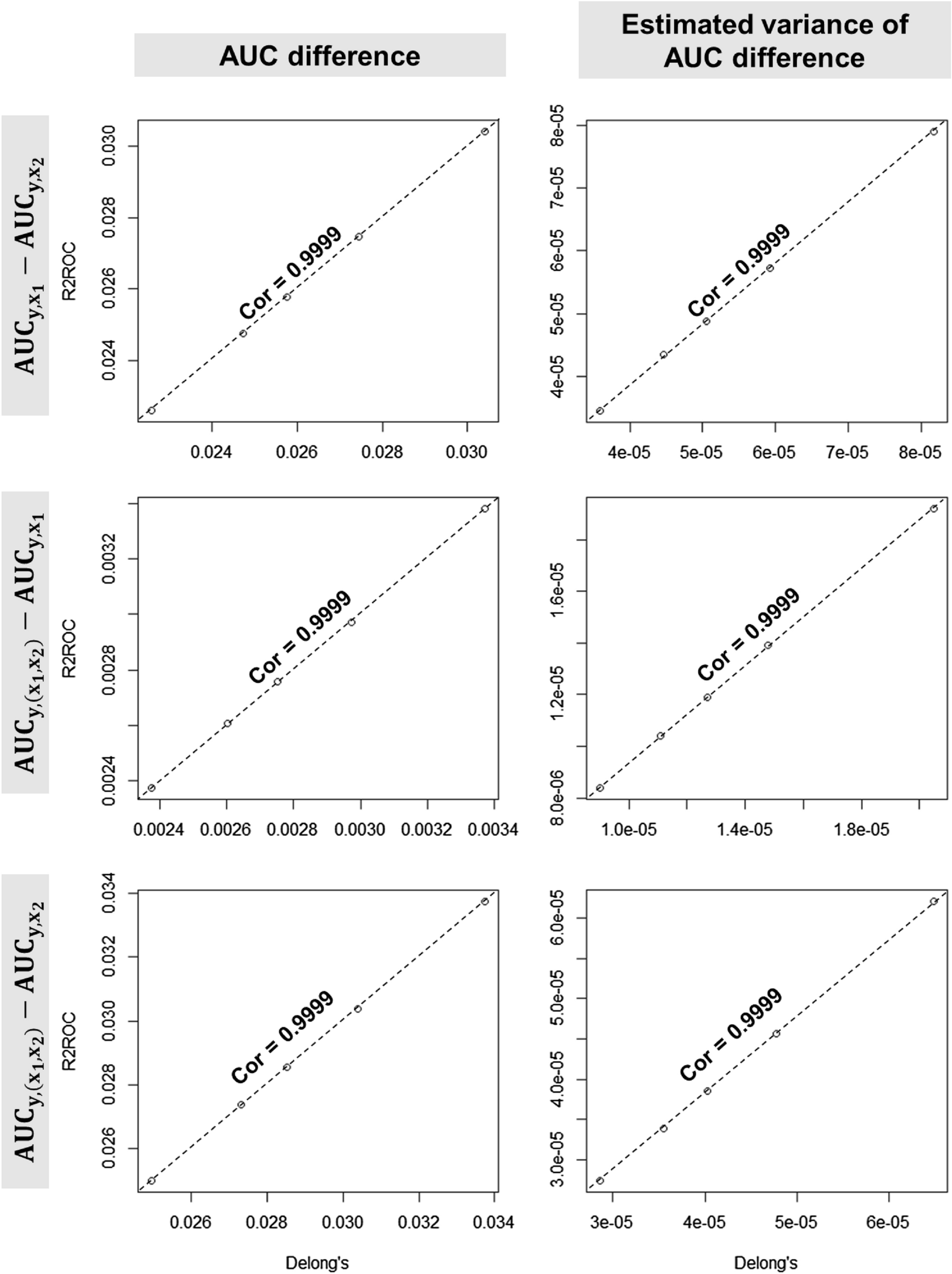
Statistical analyses were performed in Prism (GraphPad Prism 8.3) using the two-way analysis of variance (ANOVA) or students t-test. Figures were designed using Inkscape.

留言 (0)