1. IntroductionScrub typhus is a febrile illness caused by the obligately intracellular bacterium, Orientia tsutsugamushi, which is transmitted to humans via a bite of larval Leptotrombidium mites (commonly known as chiggers). Approximately 1 million scrub typhus cases occur per year in an Asia-Pacific region housing over one-third of the world’s population, termed the “tsutsugamushi triangle” [
1]. However, recent reports have indicated the presence of scrub typhus in areas previously thought free of the disease, including South America and Africa [
2,
3]. The lung is a major target organ of infection and mild interstitial pneumonia predominates in self-limiting or appropriately treated cases [
1]. However, if left untreated, disease may progress to severe lung damage and acute respiratory distress syndrome in up to 25% of cases, with a median fatality rate of 6% in untreated patients [
1,
4]. O. tsutsugamushi is a Gram-negative, lipopolysaccharide-negative coccobacillus, primarily infecting endothelial cells, monocytes, macrophages (MΦ), neutrophils, and dendritic cells [
5]. Compared with other obligate intracellular bacteria such as Rickettsia, Ehrlichia, or Anaplasma, O. tsutsugamushi has unique yet poorly understood biology [
6]. After the bacterium is internalized via endocytosis or phagocytosis, it rapidly (
5]. The bacteria utilize microtubules to traffic to the perinuclear region where replication occurs. O. tsutsugamushi replicates slowly, with peak rates occurring over 1–5 days post-infection and exits host cells via a budding mechanism [
5,
7]. A recent report has shown that O. tsutsugamushi actively inhibits NF-κΒ activation to evade host responses during its replication process [
8]. However, very few reports have examined innate recognition of O. tsutsugamushi in different lifecycle phases or mechanisms of early cytokine responses to infection. Pattern-recognition receptor (PRR)-focused investigations for O. tsutsugamushi infection are scant. While Toll-like receptor 4 (TLR4) has no direct role, TLR2 was shown to sense O. tsutsugamushi in vitro [
9]. However, in vivo experiments utilizing TLR2-deficient mice revealed a minor role in controlling bacterial growth and TLR2 was shown to promote susceptibility to lethal O. tsutsugamushi challenge via intraperitoneal inoculation [
9]. A separate study demonstrated a role for MyD88 (a key adapter protein for several TLRs), as well as cytosolic nucleic acid sensors such as RIG-I (retinoic acid-inducible gene I), MAVS (mitochondrial antiviral signaling protein), and STING (stimulator of interferon response cGAMP interactor 1), in stimulating TNFα production in infected murine embryonic fibroblasts [
10], but the biological function of these PRRs remain unclear. Likewise, the role of NLRs (nucleotide-binding site-leucine-rich repeat-like receptors) in generating inflammation has also been examined in vitro, but with conflicting results among studies [
11,
12,
13]. The role of other cytosolic sensors of infection, including Oas1-3, remain poorly understood. Additionally, whether distinct PRRs are responsible for sensing extracellular versus intracellular O. tsutsugamushi is unknown. We recently reported a significant induction of C-type lectin receptors (CLRs), including Mincle/Clec4e and Clec5a, in the lungs and brain of C57BL/6 mice lethally infected with O. tsutsugamushi, as well as a significant upregulation of Mincle in infected bone marrow-derived MΦ and neutrophils [
14]. Comparative studies of wild-type and Mincle−/− MΦ further confirmed the role of Mincle in the regulation of proinflammatory chemokines/cytokines [
14]. Mincle−/− MΦ displayed reductions in neutrophil chemoattractant (Cxcl1) and macrophage chemoattractant (Ccl2) transcription at 4 h post-infection (hpi), after bacterial endosomal escape, along with nonsignificant reductions in Tnf and Nos2. At 24 hpi, when the bacterium has begun to replicate, there was a significant reduction in type 1-skewing cytokines/chemokines (Il27, Cxcl9, Cxcl10) in Mincle−/− cells. Based on this in vivo and in vitro evidence, we proposed that Mincle upregulation contributed to the initiation and amplification of proinflammatory responses during O. tsutsugamushi infection. While this was the first report of CLR-mediated recognition of O. tsutsugamushi, the potential role of other individual CLRs or crosstalk of multiple CLRs or PRRs during infection remains unclear. The spleen tyrosine kinase (Syk) is an adaptor protein participating in the early events of CLR signal transduction [
15]. Syk-coupled CLRs are expressed predominately in myeloid cells and can be found secreted or anchored to the plasma membrane [
16,
17]. This family of receptors recognizes endogenous and exogenous carbohydrate or glycolipid moieties [
16]. CLRs interact with Syk via an immunoreceptor tyrosine-based inhibitory or immunoreceptor tyrosine-based activation motif in its own cytoplasmic tail, or through coupling with signaling partners (mainly FcγRs or DAP10/12) [
16]. The phosphorylation of Syk recruits the Malt1/Card9/Bcl10 complex, which then activates NF-κΒ or AP-1 pathways, leading to context-specific inflammatory outcomes [
16]. Given that Syk is utilized by many different CLRs, including those we have identified during O. tsutsugamushi infection (Mincle, Clec5a) [
14], those identified during Mycobacterium tuberculosis infection (Mincle, Clec5a, Dectin-1, Dectin-2, Clec4d) [
15,
18], and others, the inhibition of Syk signaling provides a useful surrogate for examining CLR pathway activation [
18,
19]. Since gene-targeted Syk deletion is lethal in vivo, numerous small molecule inhibitors including Piceatannol (PIC), R406, and BAY 61-3606 (BAY), have been commercially developed to block Syk signaling with high selectivity and specificity. Each of these inhibitors has been utilized in vitro and in vivo to deduce redundancies in CLR signaling [
20,
21,
22,
23].
In this study, we tested whether CLR-mediated signaling contributes to proinflammatory responses during O. tsutsugamushi infection. Firstly, we utilized multiple Syk inhibitors to show that bone marrow derived-MΦ from C57BL/6 mice expressed Mincle and Clec5a in a Syk-dependent fashion. Then, utilizing the selective Syk inhibitor BAY, we demonstrated that transcription of type 1 responses in MΦ relied on intact Syk signaling. Furthermore, we identified differential regulation of cytosolic innate defenses including Mx1 and Oas1-3 during infection. To our knowledge, this is the first report revealing the regulation of Mincle and Clec5a expression and the impact of their signaling during O. tsutsugamushi infection.
4. Discussion
Unlike other Gram-negative bacteria, O. tsutsugamushi lacks classical PAMPs such as lipopolysaccharide, canonical peptidoglycan, or flagellin; consequently, mechanisms of innate immune recognition have remained puzzling. This study was aimed at identifying redundancies in PRR signaling by targeting the CLR adaptor protein Syk. Using primary murine MΦ, we revealed that Mincle and Clec5a are highly transcribed and Syk-dependent during infection. We also revealed that the production of type 1 cytokines, including Il12p40, Tnf, and Il27p28, relies on intact Syk signaling. Finally, we demonstrated that infection induces transcription of cytosolic innate immune sensors Mx1, Oas1, and Oas3 in vivo and in vitro. This study is important in several aspects, suggesting that MΦ proinflammatory responses to infection are largely generated via Syk-dependent processes.
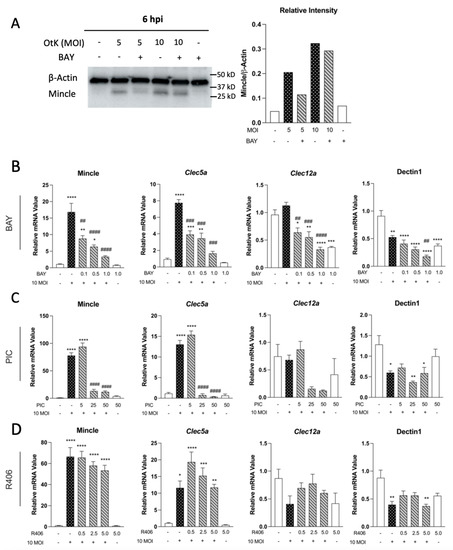
Firstly, we observed that O. tsutsugamushi infection highly upregulated Mincle in MΦ at 6 hpi, along with Clec5a, in a Syk-dependent manner (
Figure 1). In contrast, other tested CLRs were reduced (Dectin-1) or displayed no transcriptional changes (Clec12a). These findings support our previous report showing Mincle as the most highly upregulated CLR in vivo and in vitro, followed by Clec5a [
14]. Activation of Mincle, Clec5a, and Dectin-1 (among others) initiates downstream signaling via the adaptor protein Syk [
18,
33] and reports have shown this process may lead to increased CLR transcription via a feed-forward mechanism [
18]. In agreement with this notion, we observed dose-dependent decreases in Mincle and Clec5a transcription when Syk was inhibited with BAY and PIC during infection (
Figure 1). However, Mincle and Clec5a transcript levels were not completely reduced, even at the highest concentrations of inhibitors used. This indicated transcriptional regulation of these CLRs was partially reliant on Syk signaling and suggested Mincle and Clec5a may be regulated by other factors, including cytokines or DAMPs. The importance of TNF in CLR regulation was recently established, as recombinant TNF stimulation alone was shown to induce Mincle transcription and expression in MΦ [
14,
34]. Accordingly, we observed that Tnf transcription was not completely abolished, even at the highest concentration of inhibitors used (
Figure 3 and
Figure S2). Thus, it is plausible that Tnf expression via Syk-independent mechanisms stimulates the transcription of Mincle and Clec5a. We were surprised that R406 had minimal effect on the transcription of the tested CLRs. Whether this is due to differences in the selectivity of the inhibitors used or other biologic processes, such as drug metabolism, remains to be defined. Though commonly used in CLR research, Syk inhibitors display differential selectivity and potencies. PIC, a naturally occurring stilbene, was one of the first Syk inhibitors identified [
35] and arguably has been the most widely used inhibitor in CLR studies [
36,
37,
38]. However, PIC also weakly inhibits other tyrosine kinases such as Lyn, cAK, and PKC (Manufacturer Note, Sigma Aldrich). R406 is a highly selective Syk inhibitor developed for clinical use, but may also inhibit Flt3 (Manufacturer Note, Selleck Chemical). BAY is regarded as one of the most selective and potent inhibitors, with no established off-target inhibition (Manufacturer Note, Selleck Chemical). Our parallel comparison of BAY, PIC, and R406 largely confirm this notion. We found BAY treatment significantly reduced Mincle and Clec5a expression during infection at much lower concentrations than PIC or R406 (
Figure 1). Notably, the decrease in expression of Mincle and Clec5a was unlikely due to reductions in viability between the infected comparison groups (
Figure S1). This allowed us to focus on utilizing BAY for the remainder of our in vitro studies. Future efforts examining the impact of in vivo Syk inhibition with BAY could prove highly useful for examining the role of multiple CLRs in scrub typhus pathogenesis.Secondly, this study presents the first evidence for Syk-dependent MΦ responses during O. tsutsugamushi infection. MΦs play key roles in infection with O. tsutsugamushi and other closely related Rickettsia species, both as perpetrators of tissue damage and as target cells for bacterial replication [
39,
40,
41,
42]. We demonstrated here that MΦ Cxcl1 transcription, a key chemokine for neutrophil recruitment, was highly sensitive to Syk inhibition (
Figure 2). In contrast, Ccl2 (a key chemokine for macrophage recruitment) transcription was less sensitive and only reduced by ~40% at the maximal concentration of BAY. Thus, MΦ-mediated neutrophil recruitment may heavily rely on Syk signaling. This finding is important as extensive leukocyte recruitment is thought to play a prominent role in tissue damage during scrub typhus [
24,
25,
27]. Examining the effect of Syk signaling in murine models of scrub typhus via selective Syk inhibition or SYKfl/fl LysM-Cre systems is an important emphasis for future studies. Additionally, evaluating whether Syk-inhibited MΦs permit or restrict bacterial growth will shed further insight into the role of multiple CLRs controlling the infection.We were surprised to find that MΦ transcription of major type 1 cytokines, including Tnf, Il12p40, and Il27p28 were all reduced by more than 80% when Syk was inhibited during infection (
Figure 3). This is notable since our previous report utilizing single CLR-deficient (e.g., Mincle−/−) MΦs displayed nominal reductions in Tnf and Il27 transcripts early in infection (4 hpi) [
14]. While our study was limited to transcriptional analysis, our data aligns closely with a report examining type 1 cytokine production generated via J774.1 cells (a MΦ-like cell line derived from the BALB/c mouse background). These authors found inhibition of ERK, JNK, and p38 abrogated TNFα production by ~80% during O. tsutsugamushi infection [
43]. However, ERK, JNK, and p38 activation occurs multiple steps downstream of Syk activation and via multiple PRR signaling pathways. Our study permitted the evaluation of CLR-specific signaling during infection and implied a potentially important role of multiple CLRs in generating inflammation. Considering that overzealous proinflammatory/type 1 responses are thought to contribute to indiscriminate tissue damage in scrub typhus, our study presents a strong premise for future investigation into CLR engagement during disease. Thirdly, we revealed the upregulation of intracellular nucleic acid sensors Mx1 and Oas1-3 in infected mouse brains and MΦs (
Table 1,
Figure 4 and
Figure 5). Our findings corroborate a report that revealed increased expression of Oas1 and Mx1 in infected human peripheral blood mononuclear cells [
44]. Additionally, dual-RNA sequencing has uncovered a high degree of Mx1 and Oas1 gene expression in O. tsutsugamushi-infected human umbilical vein endothelial cells [
31] and a genome-wide association study of scrub typhus patients recently implicated Oas1 in susceptibility to disease [
45]. OAS family proteins sense double-stranded RNA in the cytoplasm and have been most studied in the context of viral infection [
46]. To our knowledge, only a single report has examined the contribution of OAS proteins during bacterial infection, which revealed an important role for these proteins in restricting intracellular Mycobacterium tuberculosis growth [
47]. Since Mx1, Oas1, and Oas2 were upregulated in cells that O. tsutsugamushi preferentially targets for replication (endothelial cells and MΦ), future studies utilizing knockdown or genetic deletions of these molecules are needed to delineate their role during infection. Additionally, transcription of Mx1 and Oas members has recently been shown to be regulated by multiple avenues, including both Syk-dependent [
32] and IL-27-dependent [
26] processes. We were intrigued to find that Mx1 and Oas1 transcription was reduced when Syk was inhibited, while Oas3 was not significantly impacted (
Figure 4). This indicates potentially divergent signaling pathways for the transcriptional regulation of these markers. Additionally, IL-27 stimulation prior to infection led to significantly increased Mx1 transcription in our study, but it did not significantly increase Oas1-3 transcripts (
Figure 5). This finding contrasts with a previous report, which revealed IL-27 alone in dramatically increasing Oas1 expression in the context of Zika virus infection [
26]. Whether this finding represents differences in cell types used or biological differences between target host cells of these pathogens remain unknown. Based on our findings presented herein, we propose a hypothetical model for the contribution of Syk-dependent signaling in MΦ responses to O. tsutsugamushi (
Figure 6). MΦs can sense the bacterium via Syk-dependent receptors, including Mincle, Clec5a, and potentially others, at the initial stages of infection. MΦs increase Mincle expression via feed-forward mechanisms, TNFα-mediated mechanisms, or stimulation with DAMPs as the infection progresses. Syk-dependent transcription of intracellular defense-related genes (Oas1, Mx1), neutrophil chemoattractant (Cxcl1), and Th1-skewing cytokines (Tnf, Il27, Il12) ensues. Excessive Syk activation via O. tsutsugamushi, host DAMPs, or TNFα in the microenvironment, can result in sustained inflammatory responses. These innate responses may ultimately contribute to Th1/CD8-skewed responses and acute tissue damage, as we observed in the lungs and other tested organs [
27,
48,
49]. Future investigation with targeted blockage of Syk, Mincle, or Clec5a in the host will help define the immunologic impact of CLR cooperation on scrub typhus. Such studies may help understand immune responses against other obligately intracellular pathogens, including Rickettsia, Ehrlichia, and Chlamydia, for which virtually no information is available regarding the role of CLRs in pathogen recognition.
In summary, this study provided new insight into the activation of CLR pathways for innate immune recognition of O. tsutsugamushi and identified the expression of intracellular sensors implicated in cytosolic defense. Through MΦ infection in the presence of Syk inhibitors, we provided the first evidence for Syk-dependent, type 1- and antiviral-like responses. While our studies were limited to transcriptional and predominantly in vitro data, future studies will evaluate the impact of Syk signaling in vivo. To date, PRR redundancies represent a virtually unexplored aspect of O. tsutsugamushi immunology. Considering the relatively low degree of PRR activation observed in numerous reports, it is conceivable that multiple receptors are engaged during infection. The parallel engagement of numerous PRRs may contribute to or drive overzealous immune responses. We speculate that CLR crosstalk and redundancies may contribute to malicious immune responses and progressive tissue damage in scrub typhus. A better understanding of how CLR pathway activation occurs and which CLRs are responsible for immune regulation in vivo may aid the design of treatments or vaccines for severe scrub typhus.


留言 (0)