
記住我
Human CRC cell lines (HT29, SW480, Colo205 and SW620) were obtained from the American Type Culture Collection. HT29 and Colo205 cells were cultured in RPMI-1640 medium (Invitrogen Corporation, Carlsbad, CA, USA) and SW480 and SW620 cells in Leibovitz’s L-15 medium (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 100 unit’s/mL penicillin, and 100 mg/mL streptomycin. All cultures were incubated in humidified air at 5% CO2 and 37 °C.
2.3. EV Isolation from CRC Cell LinesConfluent (70–80%) cells from all cell lines cultured in 15 cm dishes were washed twice with phosphate-buffered saline (PBS) and starved in serum-depleted medium (20 mL). After 24 h of incubation, the conditioned medium was centrifuged at 1000× g for 5 min, followed by another centrifugation step at 2000× g for 10 min, and cellular debris discarded. The supernatant was transferred to ultracentrifuge tubes for centrifugation at 10,000× g for 30 min. The medium was filtered using a 0.2 µm pore syringe filter (6786-1302, GE Healthcare, Chicago, IL, USA). A proportion of the filtered medium (1 mL) was stored for NTA and the remainder ultra-centrifuged at 100,000× g for 2 h at 4 °C using a SW32Ti rotor (Optima XE, Beckmann Coulter, Brea, CA, USA). EV pellets were washed with 10 mL PBS, followed by a second step of ultracentrifugation (SW41Ti rotor) at 100,000× g for 2 h at 4 °C. The supernatant was discarded, and the pellet resuspended in 200 µL PBS for functional assays. EVs used for protein extraction were resuspended in 250 µL of lysis buffer. The isolated EVs were stored at −80 °C for experimental use.
2.4. EV Isolation from Human Plasma SamplesPlasma samples (1 mL plasma per case) were diluted 10-fold with PBS, transferred to 50 mL tubes and centrifuged sequentially at 500× g for 5 min and 2000× g for 15 min. The supernatant was transferred to ultracentrifuge tubes and further centrifuged for 30 min at 10,000× g. The supernatant fraction obtained was collected and centrifuged for 2 h at 100,000× g. After discarding the supernatant from this step, the pellet was dissolved in PBS, passed through a 0.2 µm filter, collected in fresh tubes, and further centrifuged for 90 min at 100,000× g. The resulting pellet was resuspended in 200 µL PBS and stored at −80 °C. All centrifugation steps were performed at 4 °C using a SW41Ti rotor (Optima XE, Beckmann Coulter).
2.5. Transmission Electron MicroscopyIsolated EVs were resuspended in 4% paraformaldehyde (50–100 µL) and 10 µL aliquots were deposited on Formvar/carbon coated EM grids. The grid was covered and membrane adsorption conducted for 20 min in a dry environment. The grids (membrane side down) were transferred to drops of PBS (100 µL) with clean forceps for washing, followed by retransfer to a 50 µL drop of 1% glutaraldehyde for 5 min. Next, the grid was washed with distilled water 8 times for 2 min each, contrasted with a 50 µL drop of uranyl acetate solution for 5 min, and embedded with 50 µL methyl cellulose-UA for 10 min on ice. After removal of the grid using stainless steel loops, excess fluid was blotted and air-dried. The grid was examined under an electron microscope (JEM 1230, JEOL Ltd., Tokyo, Japan) at 80 kV.
2.6. Nanoparticles Tracking Analysis (NTA)Absolute size distribution of EVs was analyzed using Nano Sight LM10 with NTA3.2 software (Nano Sight Ltd., Minton Park, UK) for both data acquisition and analysis. Particles were automatically tracked and sized based on Brownian motion and the diffusion coefficient. Filtered PBS was used as control and blank samples. The NTA measurement conditions were as follows: temperature, 24.0 ± 0.5 °C; viscosity, 0.99 ± 0.01 cP; frames per second, 25; measurement time, 60 s. The detection threshold was similar in all samples. Three recordings were performed for each sample.
2.7. Flow Cytometry Analysis of EV-Bound BeadsEVs were attached to 4 µm aldehyde/sulfate latex beads (Invitrogen) by mixing 30 µg EVs in 10 µL beads for 15 min at room temperature with continuous rotation. The suspension was diluted to 1 mL with PBS and incubated overnight with continuous rotation at 4 °C. The reaction was terminated with 100 mM glycine and maintained for 30 min at room temperature. Next, the solution was centrifuged for 3 min at 4000 rpm in a microcentrifuge and the resulting supernatant discarded. Beads were washed twice with PBS/0.5% BSA with centrifugation at 4000 rpm for 3 min. EV-bound beads were incubated with biotin-labeled anti-CD9 (catalog number: 13-0098, eBioscience, San Diego, CA, USA) for 30 min with continuous rotation at 4 °C and rewashed twice with PBS/0.5% BSA with centrifugation at 4000 rpm for 3 min. Next, beads were incubated with streptavidin conjugated phycoerythrin (SA-PE) (BioLegend) for 30 min with continuous rotation at 4 °C. Beads were washed three times with PBS/0.5% BSA and pelleted at 4000 rpm for 3 min, and the pellet was resuspended in 1 mL PBS/BSA. Antibody-stained EV-coated beads were analyzed on a flow cytometer (FACSCalibur, BD Biosciencs, Franklin Lakes, NJ, USA) and fluorescence compared with specific antibodies and relevant isotype controls (Mouse IgG1K, eBioscience).
2.8. Western Blot AnalysisWestern blot was performed as described previously [57]. Briefly, cell lysates or isolated EV samples (containing 10 μg protein) were resolved via 12.5% SDS-PAGE, transferred to PVDF membrane, and probed using anti-CD9 (catalog number: 10626D, Invitrogen) and anti-TSG 101 (catalog number: sc-7964, Santa Cruz Biotechnology). Subsequently, blots were incubated with horseradish peroxidase (HRP) conjugated secondary antibody (1:3000) for 1 h at room temperature. Washing steps after antibody incubations were conducted on an orbital shaker four times at 10 min intervals with TTBS. Blots were developed with chemiluminescent reagent (Pierce, Waltham, MA, USA). Original blots see Supplementary File S1. 2.9. In-Solution Digestion of EVs for 2D-LC-MS/MSIsolated EVs were buffer-exchanged into 50 mM ammonium bicarbonate and prepared for analysis of protein profiles by reducing with 10 mM dithiothreitol (DTT; Merck, Darmstadt, Germany) at 56 °C for 1 h and alkylating with 30 mM iodoacetamide (IAA, Sigma, St. Louis, MO, USA) in the dark for 30 min at room temperature. After removing excess alkylating agent with 10 mM DTT at 56 °C for 10 min, protein mixtures (50 μg) were digested with 1 μg of sequencing-grade modified porcine trypsin (Promega, Madison, WI, USA) at 37 °C for 18 h. Peptide mixtures from the tryptic digestion of 50 μg EV proteins were reconstituted in 0.1% formic acid (FA; Sigma-Aldrich, Saint Louis, MO, USA), desalted using a homemade microcolumn (Source 15RPC; GE Healthcare), and analyzed via 2D HPLC coupled with a linear ion trap mass spectrometer (LTQ Orbitrap MS; Thermo Fisher, San Jose, CA, USA) operated with Xcalibur 2.2 software (Thermo Fisher).
2.10. Two-Dimensional LC-MS/MS Analysis (2D-LC-MS/MS)Dried peptides (30 µg) were reconstituted in 50 µL HPLC buffer A (30% acetonitrile/0.1% formic acid) and loaded onto a homemade strong cation exchange (SCX) chromatography column (Luna SCX 5 µm, 100 Å, 0.5 × 255 mm; Phenomenex, Torrance, CA, USA) at a flow rate of 5 µL/min for 30 min. Peptides were eluted using a gradient of 0–100% HPLC buffer B (0.5 M ammonium chloride/30% acetonitrile (ACN)/0.1% FA) and separated into 44 fractions using on-line two-dimensional high-performance liquid chromatography (2D-HPLC; Dionex Ultimate 3000, Thermo Fisher). Each SCX fraction was further diluted in-line prior to the trap of the reverse-phase column (Zorbax 300SB-C18 5 µm, 300 Å, 0.3 × 5 mm; Agilent Technologies, Wilmington, DE, USA) and diluted peptides separated on a homemade column (Synergi Hydro-RP 2.5 µm, 100 Å, 0.075 × 200 mm with a 15 μm tip; Phenomenex, Torrance, CA, USA). A linear gradient of fractionation was applied as follows: 3–28% HPLC buffer C (99.9% ACN/0.1% FA) for 37 min, 28–50% buffer C for 12 min, 50–95% buffer C for 2 min, 95% buffer C for 5 min, and 3% buffer C for 9 min at a flow rate of 0.3 μL/min. The LC apparatus was coupled with a 2D linear ion trap mass spectrometer (LTQ-Orbitrap ELITE; Thermo Fisher) operated using Xcalibur 2.2 software (Thermo Fisher). Full-scan MS was performed in Orbitrap over a range of 400–2000 Da and resolution of 60,000 at m/z 400. Internal calibration was performed using the ion signal of [Si (CH3)2O]6H+ at m/z 445.120025, 462.146574, and 536.165365 as lock masses. A total of 12 data-dependent MS/MS scan events (12 CID) were followed by 1 MS scan for the 12 most abundant precursor ions in the preview MS scan.
2.11. Data ProcessingThe raw files of resulting MS/MS spectra obtained from LTQ-Orbitrap MS were searched against 21,219 entries of Homo sapiens in the SwissProt-human database released in 20150429 using Proteome Discoverer 1.4 (Thermo Fisher). The cleaved enzyme was set to “trypsin”, with a maximum of one missed cleavage site. The precursor mass tolerance was set to 10 ppm, and fragment ions mass tolerance to 0.6 Da. Fixed modification was set to “carbamidomethylation at cysteine” and variable modifications to “acetylation at protein N-term “, “oxidation at methionine” and “deamidation at N-term glutamine”. Following the MASCOT search, the score thresholds of protein and peptide were 20 and 15, respectively. The peptide identification confidence threshold was set to “1% false discovery rate (FDR)” in the workflow of Proteome Discoverer 1.4. Peptide Validator algorithm was applied in calculation of FDR for peptide sequence analysis to distinguish true positives from random matches (decoy database).
2.12. Gene Ontology Annotation and Topological Analysis of Target ProteinsTo determine the subcellular localization and cellular compartments of the proteins involved, Human Protein Reference Database (HPRD) (http://www.hprd.org/), Protein ANalysis THrough Evolutionary Relationships (PANTHER) consortium databases (http://www.pantherdb.org/) (accessed on 8 June 2016) and Fun Rich Functional Enrichment Analysis Tool (http://www.funrich.org/) (accessed on 20 June 2016) were searched. Topological analysis of target proteins was performed using Protter (http://wlab.ethz.ch/protter/start/) (accessed on 15 September 2018), an open web application that integrates protein sequence features and transmembrane topology into illustrations [58]. 2.13. Selection of Signature Peptides and Accurate Inclusion Mass Screening (AIMS)Target proteins were subjected to in-silico tryptic digestion using MS digest software (https://prospector.ucsf.edu/prospector/html/instruct/digestman.htm) (accessed on 10 June 2022), and the signature peptides for each candidate protein selected based on the following criteria: (a) unique peptides containing 8–20 residues without known post-translational modification sites (determined from HPRD and no sequential or missed trypsin cleavage sites; (b) peptides without chemically reactive amino acids, such as C, M, and W; (c) peptides without unstable sequences, such as NG, DG, and QG; and (d) peptides without sequences potentially leading to missed cleavage, such as RP and KP. To perform AIMS, peptide samples were reconstituted in HPLC buffer A (0.1% FA), loaded across a trap column (Zorbax 300SB-C18, 0.3 × 5 mm; Agilent Technologies) at a flow rate of 0.25 μL/min in HPLC buffer A, and separated on an analytical C18 column (Synergi Hydro-RP 2.5 µm, 0.075 × 200 mm with a 15 μm tip; Phenomenex, Torrance, CA, USA). Eluted peptides were analyzed using a two-dimensional linear ion trap mass spectrometer LTQ-Orbitrap. Intact peptides were detected in the Orbitrap at a resolution of 60,000. Internal calibration was performed using the ion signal of (Si (CH3)2O)6H+ at m/z 445.120025 as a lock mass. Accurate m/z values were initially inputted into the inclusion list, and MS/MS analysis performed on the LTQ–Orbitrap MS with a 10 ppm parent mass tolerance to validate peptide detection. 2.14. EV Digestion and iTRAQ Labelling of EV Derived PeptidesEVs were isolated from plasma samples pooled from healthy controls and CRC cases using ultracentrifugation as described in Section 2.4. To EV samples (containing 50 µg protein), 1 M triethylammonium bicarbonate buffer (TEABC) was added at a final concentration of 100 mM. Next, samples were treated with 250 mM tris(2-carboxyethyl) phosphine (TCEP) to a final concentration of 5 mM. Reduced samples were incubated at 60 °C for 30 min, alkylated with IAA (10 mM), and incubated in the dark for 30 min at room temperature. Subsequently, 100 mM TEABC was added, followed by digestion with 5 µg trypsin at 37 °C for 16 h. The reaction was terminated with 1.5% FA and 0.3% trifluoroacetic acid (TFA). Digested samples were desalted using C18 beads (15 µL; LiChroprep RP-18, Merck, Darmstadt, Germany) and labeled with iTRAQ reagents (114 for HC and 116 for CRC samples) at room temperature for 2 h. Samples were desalted with C18 beads (15 µL). 2.15. 2D LC-MS/MS Analysis Coupled with iTRAQTryptic peptides (36 µg) were reconstituted in HPLC mobile phase A (30% ACN/0.1% FA) and loaded onto a homemade SCX column (Luna 5 µm SCX, 0.5 × 255 mm; Phenomenex). Peptides were eluted over a gradient of 0–100 % HPLC mobile phase B (1 M ammonium nitrate/25% ACN/0.1% FA) and separated into 72 fractions using on-line 2D-HPLC (Dionex Ultimate 3000; Thermo Fisher). Each SCX fraction was further 40-fold diluted in-line prior to the trap of reverse-phase column (Zorbax 300SB-C18 5 µm, 0.3 × 5 mm; Agilent Technologies). Diluted peptides were resolved on an analytical C18 column (Synergi Hydro-RP 2.5 µm, 0.075 × 200 mm with a 15 μm tip; Phenomenex). Linear gradient of fractionation was applied as follows: 6–9% HPLC mobile phase C (99.9% ACN/0.1% FA) for 2 min, 9–26% mobile phase C for 28 min, 26–35% mobile phase C for 10 min, 35–45% mobile phase C for 5 min, 45–63% mobile phase C for 4 min, 63–95% buffer C for 2 min, 95% buffer C for 3 min, and 6% buffer C for 5 min at a flow rate of 0.25 μL/min. The LC apparatus was coupled with a two-dimensional linear ion trap mass spectrometer LTQ-Orbitrap ELITE. Full-scan MS was performed on Orbitrap over a range of 400–2000 Da and resolution of 60,000 at m/z 400. Internal calibration was performed using the ion signal of [Si (CH3)2O]6H+ at m/z 445.120025, 462.146574, and 536.165365 as lock masses. The 12 data-dependent MS/MS scan events (6 collision-induced dissociation (CID) and 6 high energy collision-induced dissociation (HCD) modes) were followed by 1 MS scan for the 6 most abundant precursor ions in a preview MS scan.
2.16. Sequence Database Search and Quantitative Data Analysis for iTRAQData analysis was performed using Proteome Discoverer software (version 1.4, Thermo Fisher Scientific) involving the reporter ion quantifier node for iTRAQ quantification. MS/MS spectra were searched against the Swiss-Prot human sequence database (released on 20190213, selected for Homo sapiens, 20418 entries) using the Mascot search engine (Matrix Science, London, UK; version 2.5). For protein identification, the precursor mass tolerance was set to 10 ppm and fragment ion mass tolerance to 0.6 Da for the CID mode via ion trap analysis and 0.05 Da for the HCD mode by Orbitap analysis, with allowance for one missed cleavage from tryptic digestion. Fixed modification was set to methylthiolation at cysteine (+45.99 Da) and variable modification to acetylation at the protein N-terminus (+42.01 Da), oxidation at methionine (+15.99 Da), pyroglutamate conversion at N-terminal glutamine (−17.03 Da), and iTRAQ 4-plex labeling at lysine and peptide N-terminus (+144.10 Da). Based on Mascot search results, the score threshold for peptide identification was set to “1% false discovery rate (FDR)” in the processing workflow and Peptide Validator algorithm applied in calculation of FDR for peptide sequence analysis to distinguish true positives from random matches (decoy database). The decoy database was generated with Mascot with a similar size, including number of amino acids and proteins, as the original normal database. In iTRAQ quantification, each reporter ion was integrated by the mode of most confident centroid at 20 ppm tolerance, and iTRAQ-114 (114.11 Da) set as the denominator and iTRAQ-116 (116.11 Da) as the numerator for generation of a quantifiable ratio. Proteins with one unique peptide hit or without a quantifiable ratio were removed. Proteins with ratios above the mean plus one standard deviation (SD) of all ratios were classified as upregulated and those with ratios below the mean minus one SD as downregulated.
2.17. Synthesis of Surrogate PeptidesInternal standard peptides coded with stable isotope (SIS peptides) by labeling 13C/15N of Lys and Arg, leading to a 8 and 10 Da increase in the weight of peptides harboring Lys and Arg, respectively, were synthesized and purchased from Thermo Fisher Scientific. The purity of SIS peptides was >95% (in the majority of cases, purity was >98%), as determined via mass spectrometry analysis after HPLC purification. Synthetic light peptides (without stable isotope labeling) were purchased from Kelowna International Scientific (Taipei, Taiwan).
2.18. Preparation of EV Samples for MS-Based Targeted Protein QuantificationEVs were isolated as described earlier. EVs (5 μg) were dissolved in PBS containing 100 mM Tris buffer at pH 8.5. Samples were reduced with 5 mM TCEP at 60 °C for 30 min and alkylated with 10 mM IAA in the dark for 30 min at room temperature. After 2-fold dilution with 100 mM Tris, samples were digested with 0.2 μg trypsin at 37 °C for 16 h. A standard cocktail of 14 SIS peptides (containing 50 fmol 13C/15N-labeled peptides at Lys or Arg) was suspended in each EV sample followed by treatment with 0.5% FA and 0.2% TFA to acidify samples and terminate tryptic digestion. The digested samples were desalted with solid-phase extraction Oasis HLB (30 μm) cartridges (Waters, Milford, MA, USA) and lyophilized for further quantitative MS analysis using product ion scanning (PIS) mode.
2.19. LC-PIS-MS, MS Data Processing and Generation of Response CurvesDried peptides (1 µg) were reconstituted in HPLC mobile phase A (0.1% FA), loaded across a trap column (Zorbax 300SB-C18 5 µm, 0.3 × 5 mm; Agilent Technologies) at a flow rate of 20 μL/min in HPLC mobile phase A, and resolved on an analytical column (ACQUITY UPLC C18 1.7 μm, 0.1 × 100 mm) using HPLC mobile phase B (100% ACN/0.1% FA) at a flow rate of 0.4 µL/min. A linear gradient of 8–35% HPLC mobile phase B was applied in separation of the 14 target peptides for 11.3 min and a two-step 95% HPLC mobile phase B subsequently applied in regeneration to avoid carry-over into the next sample run. Eluted peptides were analyzed with a two-dimensional linear ion trap mass spectrometer (LTQ-Orbitrap ELITE). Intact peptides were detected in the Orbitrap at a resolution of 120,000. Internal calibration was performed using the ion signal of (Si (CH3)2O)6H+ at m/z 536.165365 as a lock mass. Accurate m/z values of precursor ions and their retention times were initially imported into the inclusion list. MS/MS analysis was subsequently performed on each targeted precursor and the generated product ions detected in the linear trap quadrupole (LTQ) following the scheduled scan times. The automatic gain control (AGC) value of MS and MS/MS were set to 3 × 106 ions (full scan) at 1000 milliseconds (ms) and 5 × 103 (CID) at 300 ms for maximum accumulated time or ions, respectively.
RAW files of the spectra obtained from LTQ-Orbitrap ELITE were searched against peptide sequences derived from the 14 target proteins. The SEQUEST algorithm was used for data processing and search results integrated using Proteome Discoverer 1.4 (Thermo Fisher). The MS tolerance for the monoisotopic peptide window was set to 10 ppm, and MS/MS tolerance set to 0.6 Da. Dynamic modification was set with stable isotope-containing lysine (+8 Da) and arginine (+10 Da), and charge states of the peptides set to 2+ and 3+. The false discovery rate (FDR) calculated using peptide sequence analysis to distinguish true positives from random matches (decoy database) was set to 1% as the cut-off threshold for ensuring confidence of peptide identification. Spectral libraries of sample runs constructed from PD software in msf format and relevant RAW files were imported into Skyline software (v. 21.1.0.278). Product ions with the 10 highest values of intensity were automatically selected with Skyline from ion 1 to the last ion with 1+, 2+ and 3+ ion charges in the 300–1250 m/z range and the ion match tolerance set to 0.7 m/z. After manually removing ions with interference and checking their position of retention time, the integral area of a trapezoidal model with an unsmoothed chromatogram was applied to determine the final peak area. The specific peaks of endogenous peptides were identified according to co-elution with exogenously supplemented SIS peptides and the peak areas of 2 to 7 selected fragments of target peptides summarized for further quantitative analysis. The concentration of the target peptide in samples was measured based on the ratio of the peak area to known concentrations of SIS peptides. Three independent technical repeats were performed (from digestion to the final LC-PIS-MS step) for each plasma EV sample.
Response curves for the 14 peptides were generated from experiments conducted in quintuplicate. For each target, serially diluted heavy peptide (0, 0.049, 0.098, 0.195, 0.391, 0.781, 1.563, 3.125, 6.25, 12.5, 25, 50, and 100 fmol) and a constant amount of light peptide (10 fmol) were spiked into an EV-protein digest background (1 µg protein) and analyzed via LC-PIS-MS. The MRM statistical software, QuaSAR was applied to assess the limit of detection (LOD) using the “blank and low concentration sample” method to estimate the mean of the blank samples and standard deviations of blank and low-concentration samples [59]. The lower limit of quantification (LLOQ) was calculated as the LOD value multiplied by 3 [60]. 2.20. Statistical AnalysisBar graphs for iTRAQ were generated using Sigma plot software. Data are presented as means ± standard deviation. The Mann–Whitney test was used to compare the differences in plasma derived EV levels of selected proteins between HC and CRC groups. Mann–Whitney and Kruskal–Wallis tests were employed to evaluate the association of plasma-EV ADAM10, CD59, TSPAN9 and CEA with various clinicopathological parameters of CRC patients. The diagnostic power of ADAM10, CD59, TSPAN9 and CEA was analyzed by constructing a receiver operating characteristic (ROC) curve with sensitivity versus 1-specificity and calculating the area under the ROC curve (AUC). For all statistical analyses, a two-tailed p-value ≤ 0.05 was considered significant. Calculations and diagrams were generated using GraphPad Prism 7.0 (GraphPad Software, Inc., San Diego, CA, USA).
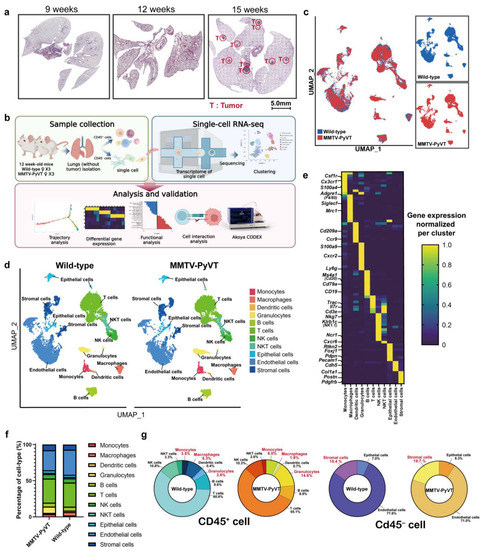
4. DiscussionIt is well recognized that EVs carry many membrane proteins on their surface that can transduce signals to recipient cells and serve as biomarkers and target proteins for the diagnosis and treatment of diseases [61,62]. A number of EV membrane proteins, including CD147/basigin and EpCAM, have been selected to pair with the EV markers CD9 or CD63 as biomarker panels for CRC and have been successfully tested for effective cancer detection using blood specimens [40,41]. These promising findings prompted the present effort to discover novel CRC plasma EV biomarkers, with a focus on membrane proteins. Through in-depth analysis of EV membrane protein profiles from CRC cell lines and an iTRAQ quantitative analysis of differentially expressed plasma EV proteins from CRC patients and HCs, we generated a biomarker candidate list. We further prioritized the candidate targets by AIMS-based detection of all possible candidates in plasma EVs from CRC patients and HCs, and successfully verified the prioritized candidate targets by targeted MS-based absolute quantitative analysis of prioritized candidates in individual plasma EV samples from age/sex-matched CRC patients and HCs. Finally, we generated a novel two-marker panel consisting of EV membrane proteins CD59 and TSPAN9 that effectively discriminated CRC patients from HCs (AUC = 0.98). To our knowledge, this is the first study to apply targeted MS for verification of multiple (in our case, more than a dozen) EV membrane proteins as plasma cancer biomarkers in which the amounts of target proteins were quantitatively measured in individual plasma EV samples by spiked SIS peptides with known quantities. Thus, for the first time, we were able to simultaneously measure the absolute levels of multiple EV membrane proteins in individual plasma EV samples and perform head-to-head comparisons of selected targets’ ability to detect CRC from the same clinical sample set. Such a targeted MS-based biomarker verification pipeline has been successfully applied to identify promising body fluid-accessible biomarkers or biomarker panels for detection of cancer and other diseases [63,64,65,66,67]. Candidate proteins that stand out from the remaining targets in this kind of head-to-head comparison have a better chance of surviving and becoming clinically applicable biomarkers during further validation testing using samples from a large cohort. For example, we recently validated saliva matrix metalloproteinase 1 (MMP-1) as a strong diagnostic marker of oral cavity cancer in saliva samples from a large cohort (n = 1160), supporting previous reports that this protein is one of the most promising salivary biomarkers for oral cavity cancer using a targeted MS-based assay to compare multiple candidate proteins [66,68].Advances in MS-based proteomics methods have greatly enhanced the biomarker discovery and verification pipeline through targeted and non-targeted approaches. Proteome cataloguing of EVs from various cancer types has revealed numerous membrane and cytosolic proteins, as well as a set of proteins distinct for different types of malignancies, that reflect the original host cell. For the most part, discovery efforts have focused on tissues, proximal fluids or cell lines, with plasma being used only after candidates have been found. However, there are no reliable principles for determining which tissue or cellular proteins will work as plasma biomarkers, and the error rate is considerable. In general, clinical data are rarely used in discovery attempts to select markers that will have the best chance of success in the targeted clinical environment. As a result, discovery efforts generate vast lists of candidate biomarkers, but they also allow for the testing of the greatest number of biomarkers possible, increasing the chances of finding clinically meaningful indicators.
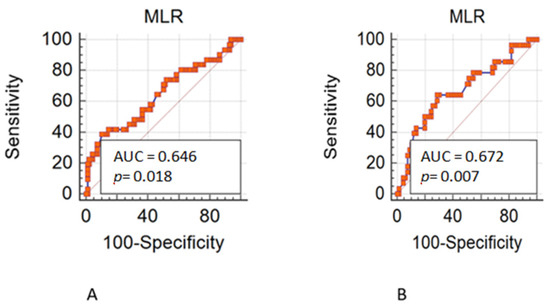
AIMS, implemented before committing to the time and resource-intensive procedures of developing a quantitative test, allows for prioritizing of a large number of biomarker candidates based on their detection in plasma. It is a remarkable technique for bridging the gap between unbiased discovery and MS-based focused assay creation [51]. Following this approach, we employed AIMS as an empirical prioritizing step to determine whether biomarker candidates could be detected in plasma and additionally used iTRAQ to validate the targets detected by AIMS (Figure 4). Among the 13 prioritized targets, eight (ADAM10, ALCAM, CD58, CD59, ITGAM, SELP, TSPAN9 and TTYH3) were previously reported to be dysregulated in tissue and/or plasma specimens from CRC patients [69,70,71,72,73,74,75,76]. Even though many of the selected candidates had been reported as potential markers for CRC, only three targets (ADAM10, CD59 and TSPAN9) showed good power (AUC > 0.8) to discriminate CRC patients from HCs in the current study (Table 2 and Figure S4). Our findings echo previous observations regarding the high error rate for verification of liquid biopsy biomarkers and support the need for further clinical validation of verified biomarkers/biomarker panels. CD59, a GPI-anchored membrane protein, inhibits the development of the membrane attack complex, preventing complement activation and perhaps protecting cancer cells from complement-dependent cytotoxicity [77,78]. Recent studies have indicated that CD59 is highly expressed in several cancer cell lines and tumor tissues, including those from CRC patients [78]. It has been shown that expression levels of membrane-bound complement regulatory proteins (mCRPs), including CD46, CD55 and CD59, are considerably higher in colon cancer tissues than in normal neighboring normal colon tissues. Furthermore, an analysis based on TNM staging showed that the expression of CD55 and CD59 is higher in stage III and stage IV colon cancers than in stage I and stage II tumors [79]. Another study that applied tissue microarrays to evaluate the immunohistochemical expression of CD59 in over 460 well characterized CRC cases similarly found an association between high CD59 expression and worse prognosis, a relationship that held particularly well for patients in the early stages of the disease [71]. CD59 has also been reported to be overexpressed in several other cancers, including breast, esophageal and pancreatic cancer, and that its expression may facilitate tumor cell escape from complement surveillance [80,81,82,83]. However, to date no studies on the pathobiological role of CD59 in EVs or its potential clinical utility in cancer detection has been reported. Tetraspanins are a vast superfamily of glycoproteins whose tetraspanin-enriched microdomains engage in a variety of specific molecular interactions. Among the tetraspanins previously identified on a variety of EVs are CD9, CD63, and CD81 [84]. We found 11 tetraspanins (TSPAN1, TSPAN5, TSPAN6, TSPAN8, TSPAN9, TSPAN14, TSPAN15, CD9, CD81, CD82, and CD151) in the EV proteome of at least three CRC cell lines and detected CD63 in the EV proteome of Colo205 and SW620 cell lines (Tables S2 and S3). TSPAN9, a 4-transmembrane protein involved in tumor growth and signal transduction, has been shown to be linked to tumor invasion, metastasis, and autophagy. The understudied gene encoding this tetraspanin was reported to be amplified in serous fallopian tube cancer [85]. Previous studies also revealed that high expression of TSPAN9 is an independent prognostic factor for poor overall survival of patients with gastric cancer [86] and showed that TSPAN9 is overexpressed in 5-fluorouracil (5-FU)-resistant gastric cancer cells, where it contributes to the chemoresistance to 5-FU by promoting autophagy [87]. However, there has been no research on the expression of TSPAN9 in human CRC tissue or tissue-derived EVs. In this context, we are also the first to explore the clinical significance of plasma EV levels of CD59 and TSPAN9 in cancer. In addition to identification of plasma EV CD59 and TSPAN9 as a two-marker panel for CRC detection, the pathobiological roles of EV CD59 and TSPAN9 in the tumor microenvironment and/or chemoresistance of CRC obviously remain as intriguing issues that warrant further investigation.Through in-depth analysis of EV protein profiles from four CRC cell lines and bioinformatics annotation of the identified proteins, we have generated a focused EV membrane protein dataset containing 518 ‘core’ proteins released in common from at least three CRC cell lines (Tables S2 and S3). We found that 16.4% (754/4586, HT29), 20.5% (666/3243, SW480), 22.9% (503/2196, SW620) and 15.8% (780/4924, Colo205) of the identified EV proteins were classified as membrane proteins (Figure 3). The average across cell lines was 18.9%, which is close to the estimated value obtained from an analysis of proteomic data collected from the Vesiclepedia database in which 627 of 3027 evaluable EV proteins (21%) were classified as plasma membrane proteins [88]. By combing the EV proteome MS dataset with topological information on EV membrane proteins obtained by analysis using Protter software, we were able to map the location(s) of MS-detected peptide(s) in the amino acid sequences of three types of membrane proteins: single-transmembrane domain-containing proteins, multiple-transmembrane domain-containing proteins, and anchored proteins (Figure 3D and Table S4). This information is very useful for designing workable targeted MS assays for detection and quantification of these EV membrane proteins in different biological or body fluid samples from distinct diseases or under different experimental conditions. Our study used differential ultracentrifugation to isolate EVs from cell conditioned medium and plasma, which has its own lacunae. Although ultracentrifugation can yield significant quantities of highly pure exosomes, it is unsuitable for clinical diagnosis owing to its low throughput, which only allows for the identification of six samples at a time, and poor reproducibility, making study-to-study comparability suspect. Ultracentrifugation paired with density gradient centrifugation (DGC) can improve exosome purity; however, DGC is time-consuming (centrifugation for 16–90 h), limiting its usefulness in clinical settings. A recent development of a magnetic bead-based EV enrichment approach for automated and high-throughput processing of body fluid samples in 96-well plates may represent an alternative purification strategy to achieve high throughput [89]. Another aspect is to increase the sample size to obtain more reliable and accurate mean values, uncover outliers that could bias data in a smaller sample, and achieve a lower margin of error. Furthermore, the inclusion of a maximum number of candidates in analysis pipelines can increase the likelihood of discovering therapeutically important indicators. In future studies, we would seek to increase case study numbers and include more targets for verification from our 2D-LC MS/MS and iTRAQ findings and possibly use a more sensitive and higher throughput MS assay, such as multiple-reaction monitoring (MRM) or parallel reaction monitoring (PRM) MS.
留言 (0)