Cell lines and reagents
The lung cancer cell lines H460 control and H460-shRab37 knockdown cells were established from parental H460 cells and the stable lung cancer cell lines expressing active-form Rab37 (Q89L) and vector control transgenes were established from parental CL1-5 cells by Dr. Yi-Ching Wang (National Cheng Kung University, Tainan, Taiwan). The stable cell lines CL1-5-Rab37-Q89L-shGFP, shAtg5, shAtg7, and shSec22b cells were established by infection with lentivirus containing shRNAs (Academia Sinica, Taipei, Taiwan) followed by selection with puromycin (Sigma, MO, USA). All cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, NY, USA) with 10% fetal bovine serum (Gibco) and penicillin/streptomycin (Sigma) at 37 °C in a 5% CO2 incubator. Amiodarone, niclosamide, and spautin-1 were purchased from Sigma, MO, USA).
GTP-agarose pulldown assay
Cells were treated under different conditions and followed by protein extraction. Protein lysate was incubated with ActivX Desthiobiotin-GTP probe™ (Thermo Scientific, IL, USA) overnight at 4 °C. The beads were centrifuged and washed twice with GTP-binding buffer [50 mM Hepes (pH 7.4), 1% Triton X-100, 10% glycerol, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, and 1% protease inhibitor cocktail]. The GTP-bound proteins were resuspended with SDS gel loading buffer and the GTP-Rab37 protein expression was detected using anti-Rab37 antibody by immunoblotting.
Immunofluorescent staining
The cells were seeded onto a 2-well chamber slide (SPL, Korea) and treated with different conditions. Anti-TIMP1 (ab109125, Abcam), anti-LC3 (PM036, M186-3, MBL), or anti-Rab37 (LTK BioLaboratories, Taiwan) antibody was used. The fluorescent change of the cells was detected under a multi-photon confocal microscope (Olympus, FV1000MPE, Tokyo, Japan). A detailed description is provided in our previous report [29].
Immunoblotting
The total protein of the cell lysates was extracted after treatment with different conditions. A detailed description of the preparation is presented in our previous report [29]. The primary antibodies used to detect specific proteins were β-actin (A544, Sigma), LC3 (PM036, Medical and Biological Laboratories, Nagoya, Japan), TIMP1 (ab109125, Abcam, Cambridge, UK), Rab37 (LTK BioLaboratories, Taiwan), ATG5 (ab108327, Abcam), and ATG7 (ab133528, Abcam). Samples were incubated overnight at 4 °C. The membranes were incubated with the secondary anti-rabbit (Amersham Pharmacia, Piscataway, NJ, USA) or anti-mouse (Chemicon, Temecula, CA, USA) antibody at room temperature for 1 h. Finally, the membrane was rinsed with enhanced chemiluminescence (ECL) (WBKLS0500; Millipore) and exposed using the BioSpectrum AC system (101-206-009; UVP, Upland, CA, USA).
Immunoprecipitation
The extracted protein (1 mg) and beads were incubated with LC3 or IgG antibody. This mixture was rolled at 4 °C overnight. Beads were collected by centrifugation at 10,000 rpm for 1 min at 4 °C followed by extensively washing with PBS buffer and resuspending with SDS gel loading buffer. The protein samples were analyzed by immunoblotting.
Preparation of conditional medium (CM)
Cells were cultured in a 6 cm dish with 4 ml serum-free medium for 24 h to prepare the conditional medium. The media were collected using Amicon Ultra centrifugal filter units (UFC900396, Millipore) according to the manufacturer’s protocol, and the total protein concentration in conditional medium samples was determined by Bradford assay (Bio-Rad Laboratories Inc.).
Transmission electron microscopy (TEM)
The cell and autophagosome fraction (AP) were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer at 4 °C for 10 min. A detailed account of the procedure for sample preparation is described in a previously published study [29]. The samples were then sectioned, and the sliced samples were investigated under a TEM (Hitachi, Japan, HITACHI-7000).
Negative staining of the purified autophagosome by immunogold labeling
In brief, 4 μl of purified autophagosome was loaded onto the carbon film supported nickel grid (CF400 Ni, EMS Microscopy Academy). After 1 min, the sample was removed by Whatman filter paper. LC3 protein on the surface of the autophagosome was recognized by the primary antibody (M152-3, MBL), which was diluted 50-fold in HB buffer. After 1 h, 10-nm gold immunogold-conjugated antibody (Abcam), which was diluted 50-fold in HB buffer, was used to detect the primary antibody. Finally, the grid was washed once by ddH2O, and the sample was negatively stained with 2% uranyl acetate for 1 min. The samples were investigated under a TEM (JEOL, USA, JEM-1400) at 120 kV with a Gatan Model 895 digital camera.
Total internal reflection fluorescence (TIRF) imaging
Cells received the same treatment as immunofluorescent staining. A TIRF system was equipped with a high-sensitivity EMCCD Camera (iXOn3897, Andor technology) and an UPON 100XOTIRF objective lens (NA = 1.49; Olympus). We tracked each vesicle trafficking distance with track IT software (Olympus) and a thickness of 100 μm.
Autophagosome purification
Cells (in 10% sucrose) were mixed with 0.5 ml of the buffer (1 M Hepes/0.1 M EDTA) and homogenized using a Dounce homogenizer. This homogenate was diluted with homogenization buffer (HB; 0.25 M sucrose, 10 mM HEPES, 1 mM EDTA, pH 7.3) containing 1.5 mM glycyl-l-phenylalanine 2-naphthylamide (GPN) and 1% DMSO, to a final GPN concentration of 0.5 mM and a final homogenate concentration of 5%. After incubation for 7 min at 37 °C to destroy the lysosomes, the homogenate was cooled to 4 °C. The tubes of GPN-treated homogenate were centrifuged at 4000 revolutions per minute (rpm) for 2 min to collect the supernatant, which was placed on top of a discontinuous (two-step) Nycodenz gradient, prepared by diluting isotonic (36% w/v) Nycodenz with HB to obtain a top layer of 9.5% Nycodenz and a bottom layer of 22.5% Nycodenz. The Nycodenz gradient was centrifuged overnight at 26,000 rpm in an SW28 rotor (Beckman, USA), and the gradients were divided into three fractions. The interface band was diluted with HB and layered on top of a discontinuous gradient of 33% Percoll in HB on top of 22.5% Nycodenz in HB, and centrifuged for 1 h at 20,000 rpm in the SW28 rotor. Autophagosomes formed a band at the lower interface, from which it could be recovered for analysis. Percoll was removed from the fractions of interest (e.g., the autophagosomes) by mixing with isotonic 60% (w/v) iodixanol in water, and centrifuging for 30 min in an SW40 rotor at 20,000 rpm. The autophagosome band was collected from the interface band [29].
In solution digestion and LC–MS/MS analyses
The protein was extracted from the autophagosomes of CL1-5 cells. Protein extracts were denatured and then alkylated in dithiothreitol (7 mM) and iodoacetamine (21 mM), respectively, at 37 °C for 30 min. Proteins were digested by trypsin at 37 °C for 16 h. The peptide mixtures were separated on a 3 × 150 mm C18 column (Gemini, Phenomenex, Torrance, CA, USA) coupled to a high-performance liquid chromatography system (Beckman Coulter, CA, USA) using an acetonitrile gradient in 0.1% ammonium hydroxide solution, with about 30 fractions. The peptide fractions were analyzed on a nanoLC-Q Exactive™ HF mass spectrometer (Thermo Fisher, San Jose, USA) equipped with an HPLC system (M Class, Waters, MA, USA). MS raw files were uploaded into Proteome Discoverer (version 2.1, Thermo Fisher, MA, USA) with the default setting to generate peak lists for protein identification using the MASCOT search engine (version 2.5, Matrix Science, MA, USA) against the Swiss-Prot Mus musculus protein database (released in Jan 2016). The peptides sharing an identical sequence among multiple proteins were assigned to the one with the highest protein score. The identification of peptides and proteins with a false discovery rate of less than 1% was considered acceptable.
Lung-to-lung metastasis assay
Male NOD/SCID mice (6–8 weeks old) were obtained from the Laboratory Animal Center of National Cheng Kung University (Tainan, Taiwan). The experimental protocol complied with Taiwan’s Animal Protection Act and was approved by the Laboratory Animal Care and Use Committee of National Cheng Kung University. A total of 5 × 104 cells were suspended in 100 ml matrigel (BD Biosciences) in serum-free DMEM. Mice were anesthetized by intraperitoneal administration of Zoletil 50 solution (10 mg/ml, Virbac, Taipei, Taiwan) combined with 2.3 mg/ml of Xylazine Hydrochloride (also named Rompun) (Bayer Leverkusen, Germany) at a dose of 0.8 to 1 mg/10 g of body weight. After mice had been anesthetized with Zoletil 50 solution, they were placed in the left lateral decubitus position. Tuberculin syringes (10 μl) with 23-gauge hypodermic needles were used to inject the cell inoculum percutaneously into the right lateral thorax, at the lateral dorsal axillary line, 1.5 cm above the lower rib line, just below the inferior border of the scapula. The needle was quickly advanced 5–7 mm into the thorax and was slowly removed after the injection of cell suspension. After the tumor injection, the mouse was turned to the right lateral decubitus position. Animals were observed for 45–60 min until fully recovered.
Immunohistochemistry (IHC)
The slides were prepared as described in our recent report [29]. The primary antibody was used to detect TIMP1 (ab109125, Abcam). The Dako REAL Envision Detection system (Dako, Glostrup. Denmark) was used to detect the protein expression. Hematoxylin (Merk, Darmstadt. Germany) was used to demonstrate nuclear morphology, and DAB (Dako) was used in conjugation with immunoperoxidase detection systems.
Statistical analysis
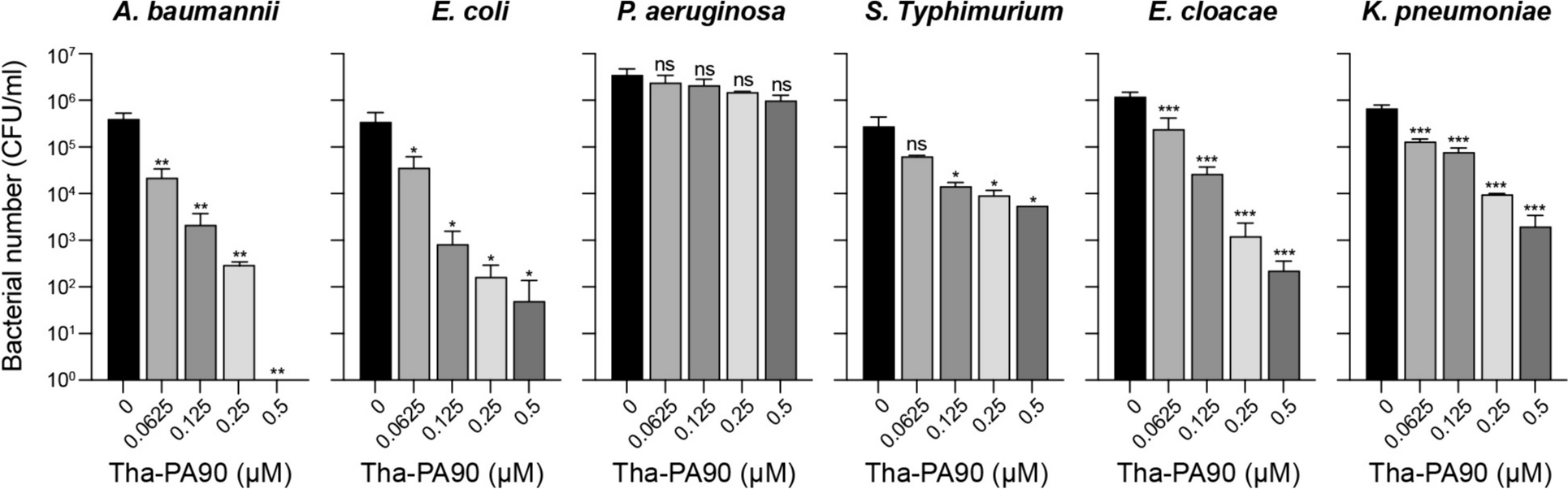
Data are presented as the mean ± standard deviation (SD) values (error bars). Differences between the experimental and control groups were analyzed by two-tailed Student’s t-test. P value was considered statistically significant: *p < 0.05, **p < 0.01, and ***p < 0.001.





留言 (0)