
記住我
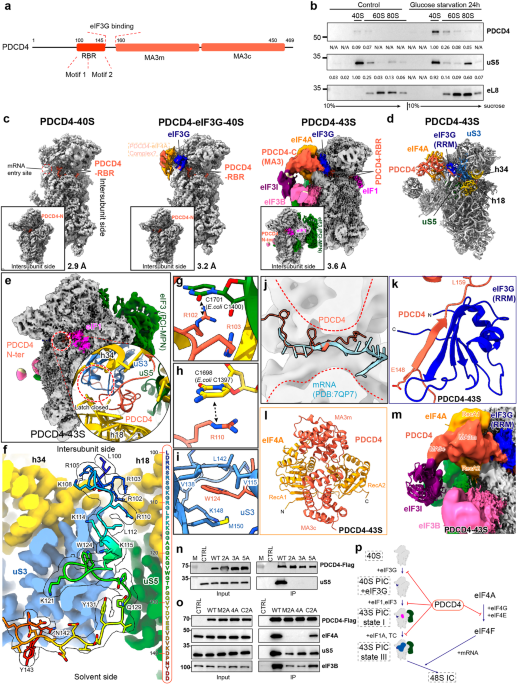
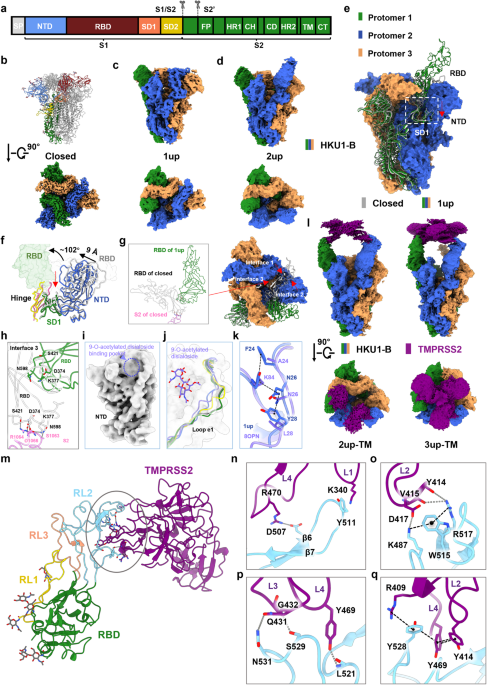
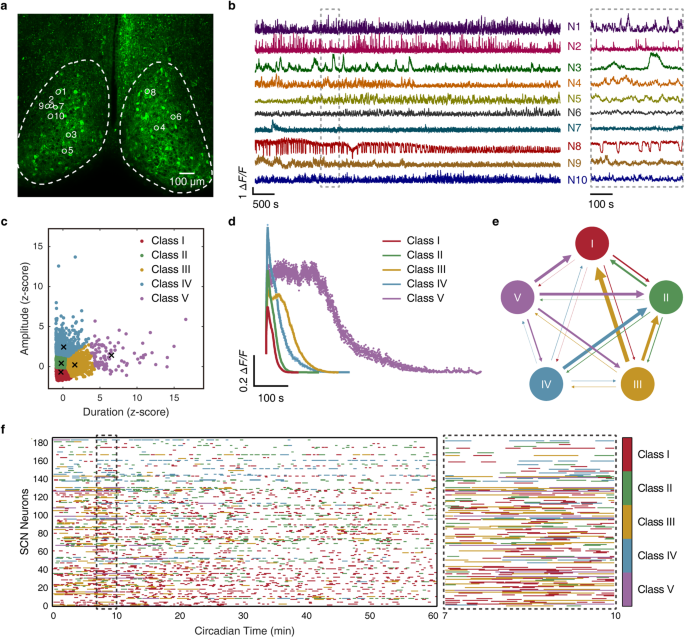

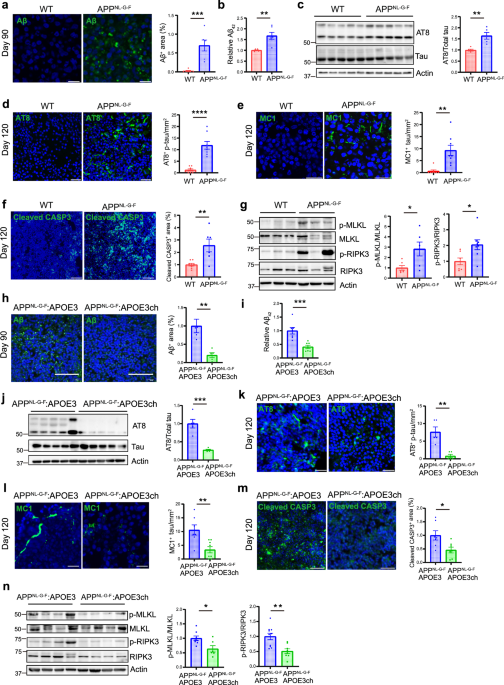
All human cancer tissues were obtained from the Department of Gastrointestinal Surgery and Department of Hepatological surgery, the First Affiliated Hospital of Sun Yat-sen University (IRB: 2017247) or the Department of Surgery, the Affiliated Hospital of Nanjing University (Nanjing Drum Tower Hospital, IRB: 2020-072-01). Tissues were obtained with patients’ written consent under a protocol approved by the institution’s Institutional Review Board and confirmed by a pathologist before use.
Cell lines and cell cultureAll human cell lines were obtained from ATCC (BxPC-3: CRL-1687; Capan-2: HTB-80; Panc02.03: CRL-2553; CFPAC1: CRL-1918; PANC-1: CRL-1469; AsPC-1: CRL-1682; and HEK293T: CRL-3216). Cells were maintained at 37 °C in a humidified atmosphere with 5% CO2. Cells were cultured in either RPMI 1640 or Dulbecco’s modified Eagle’s medium (DMEM) according to ATCC instructions with 10% fetal bovine serum (FBS). RAS-less MEF cell lines stably overexpressing KRASG12D or KRASG12V were obtained from NIH’s RAS Initiative and cultured as indicated in the instructions online (https://www.cancer.gov/research/key-initiatives/ras/ras-central/blog/2017/rasless-mefs-drug-screens). The in-house primary KRASG12D MEF cell line was established from E13.5 LSL-KrasG12D genetic mouse and cultured in DMEM with 10% FBS and 1% GlutaMAX (Thermo Fisher, MA, USA, 35050061). Two additional cell lines were used to produce condition medium for PDAC organoid culture. L Wnt-3A (CRL-2647™) was purchased from ATCC for Wnt3a conditioned medium production and cultured in advanced DMEM/F12 supplemented with 10% FBS. HA-R-Spondin1-Fc 293T Cells (R&D, Minneapolis, USA, 3710-001-01) for producing RSPO-1 conditioned medium was cultured in DMEM supplemented with 10% FBS. All cell lines were regularly tested for mycoplasma contamination.
Ribosome sequencing and data processingThe ribosomal profiling technique was processed as reported previously,34 with a few modifications as described below. Cells or tissues were treated with harringtonine (2 µg/mL, Abcam, Waltham, USA, ab141941) for 2 min and cycloheximide (100 µg/mL, Sigma, MO, USA, 66-81-9) for 3 min. Samples were then dissolved in lysis buffer. Lysates were centrifuged at 20,000× g for 10 min at 4 °C, then the supernatant was incubated with 7.5 µL RNase I (NEB, Ipswich, USA, M0307) and 5 µL DNase I (NEB, Ipswich, USA, M0303) in 300 µL total volume for 45 min at room temperature followed by the addition of 10 µL SUPERase·In RNase inhibitor (Ambion, TX, USA, AM2696) to stop nuclease digestion. Size exclusion columns (Illustra MicroSpin S-400 HR Columns, GE Healthcare, 27-5140-01) were equilibrated with 3 mL of polysome buffer by gravity flow and then centrifuged at 600× g for 4 min at room temperature. 100 μL of digested RPFs was then added and centrifuged at 600× g for 2 min. 10 μL of 10% (wt/vol) SDS was then added and RPFs with size greater than 17 nt were isolated according to the RNA Clean and Concentrator-25 kit (Zymo Research, Irvine, USA, R1017). rRNA was removed using the method reported previously.35 In brief, short (50–80 bases) antisense DNA probes complementary to rRNA sequences were added to solution containing RPFs; then RNase H (NEB, Ipswich, USA, H0110) and DNase I was added to digest rRNA and residual DNA probes. Finally, RPFs were further purified using magnet beads (Vazyme, Nanjing, China, N412). After obtaining RPFs, Ribo-seq libraries were constructed using NEB Next® Multiple Small RNA Library Prep Set for Illumina® (E7300S, E7300L). In brief, adapters were added to both ends of RPFs, followed by reverse transcription and PCR amplification. The 140–160 bp size PCR products were enriched to generate a cDNA library and sequenced using Illumina HiSeqTM X10 by Gene Denovo Biotechnology Co. (Guangzhou, China).
RNA sequencing and data processingTotal RNA was first extracted using Trizol reagent kit (Invitrogen, Carlsbad, CA, USA, 15596026) according to the manufacturer’s protocol. Eukaryotic mRNA was then enriched by Oligo(dT) beads and fragmented into short fragments using fragmentation buffer and reverse transcribed into cDNA with random primers. Then the cDNA fragments were purified with QiaQuick PCR extraction kit (Qiagen, Germantown, USA, 28104), end-repaired, poly(A) added, and ligated to Illumina sequencing adapters. The ligation products were size selected by agarose gel electrophoresis, PCR amplified, and sequenced using Illumina HiSeq2500 by Gene Denovo Biotechnology Co. (Guangzhou, China).
Plasmid construction and transfection of HEK293T cellsRASON, NF1, LINC00673, LINC00673-ORF, LINC00673-ORF-ATG-MUT, and KRAS expression plasmids were cloned into the pCDH-CMV-MCS-EF1-GFP + Puro vector (SBI, CA, USA, pCD513B-1). Plasmids were transfected into cells using Lipofectamine 3000® (Thermo Fisher, MA, USA, L3000015) according to the manufacturer’s protocol.
Stable transfection of cancer cells using lentiviral vectorsLentiviral vectors expressing human RASON, mouse Rason, sh-RASON, KRAS, sh-KRAS, or sh-LINC00673 were co-transfected with packaging vectors psPAX2 (Addgene, MA, USA, 12260) and pMD2G (Addgene, MA, USA, 12259) into HEK293T cells for lentivirus production using Lipofectamine 2000 in accordance with the manufacturer’s instructions. To establish stable cell lines, cancer cells were transduced using the respective lentivirus in the presence of polybrene (8 mg/mL, Sigma, MO, USA, TR-1003-G). After 72 h recovery, cells were selected with 2 mg/mL puromycin.
CRISPR-mediated RASON KO in human PDAC cell linesThe target sequences of RASON sgRNA (TTGGATGGAAAGTGGGGAAT) were designed using the http://crispr.mit.edu/ online tool. To produce CRISPR-lentivirus, HEK293T cells seeded in 100 mm plates were transfected with 10 μg lentiCRISPRv2-gRNA or lentiCRISPRv2 control (Addgene, MA, USA, plasmid #52961) plasmids, 5 μg psPAX2 and 2.5 μg PMD2G plasmids using Lipofectamine 3000 according to the manufacturer’s instructions. After incubation for 48–72 h, the supernatants containing lentivirus were harvested and used to infect cells for 4–6 h. Polyclonal KO cell lines were harvested after five days. Monoclonal cell lines with stable KO of target gene were selected over 1−2 months in 96-well plates.
CRISPR-mediated mouse Rason KO in Ras-less MEF cellsThe sgRNA sequences targeting mouse Rason (CGCGGATTGTGTGTTTGCGT and GGGTTGGGGTTCCCCTAACG) were cloned into the eSpCas9-2A-GFP (PX458) plasmid by clone EZ. Plasmid amplification was carried out using DH5α competent cells. Ras-less MEF cells were seeded in 100 mm dishes and transfected at approximately 85% confluency. In brief, 30 μg of sgRNA expression plasmids were transfected using Lipofectamine 3000 according to the manufacturer’s protocol. Three days later, GFP-positive cells were sorted into 96-well plates by flow cytometry, single cell clones were picked and validated after 2 weeks.
CRISPR-mediated NF1 KO in Ras-less MEF cell linesThe sgRNA sequences targeting mouse NF1 (CCAGGACATCTCCAAGGATG) was cloned into pLentiCRISPR v2. KRASG12D MEF cells were seeded in 100 mm dishes and transfected at approximately 85% confluency, followed by puromycin selection.
PCRDNA was extracted with DNeasy Blood &Tissue Kit (Qiagen, Germantown, USA, 69504) according to the standard procedure, and subjected to PCR assay. Primer sequences are: mouse Rason (forward, 5′-CTTCTGTTTCGGGCTGTACG-3′; reverse, 5′-GGCCAATACCCATCTCTCCA-3′). The resulting DNA was evaluated by agarose gel electrophoresis followed by ethidium bromide (Thermo Fisher, MA, USA, 15585011) staining.
Reverse transcription and real-time (RT) PCRTotal RNA was extracted with TRIZOL and cDNA was obtained by using PrimeScript RT Master Mix (Takara Bio, Kusatsu, Japan, RR036A). The resulting cDNA was then subjected to RT-PCR analysis with SYBR Select Master Mix (Thermo Fisher, MA, USA, A46012) in a StepOnePlus realtime PCR system (Applied Biosystems). Results were normalized to β-Actin mRNA in each sample. Primer sequences are: LINC00673 (forward, 5′-AGTCTGGAGCGCAGAGGACA-3′; reverse, 5′-TCAATCCACGGATGGAGAAGAG-3′), β-Actin (forward, 5′- CATGTACGTTGCTATCCAGGC-3′; reverse, 5′- CTCCTTAATGTCACGCACGAT-3′).
Antibody generation and immunoblottingA rabbit monoclonal antibody against the N-terminal 1–14 residues of human RASON protein was obtained by inoculating rabbits with synthesized peptides. The antibody was purified using affinity chromatograph columns. Cell or tissue lysates were separated by 7.5%–17% SDS-PAGE and subjected to western blotting analysis using the following primary antibodies: anti-RASON (This study; 1:500), anti-C-RAF (CST, Danvers, USA, Cat#9422; 1:1000), anti-p-C-RAF (CST Cat#9421; 1:1000), anti-ERK1/2 (CST Cat#4695; 1:1000), anti-p-ERK1/2 (CST Cat#4370; 1:1000), anti-AKT (CST Cat#4691; 1:1000), anti-p-AKT S473 (CST Cat#3787; 1:1000), anti-p-AKT T308 (CST Cat#13038; 1:1000), anti-β-actin (Sigma, MO, USA, Cat#SAB1305567; 1:5000), anti-FLAG (Sigma Cat#SAB1306078; 1:1000), anti-KRAS (Abcam, Waltham, USA, Ab180772; 1:1000), anti-NF1 (Abcam Ab17963; 1:1000), anti-Rho A + B + C (Abcam Ab175328; 1:1000), anti-cdc42 (Abcam Ab187643; 1:1000), anti-ARHGAP10 (Abcam Ab222805; 1:1000). HRP-labeled secondary antibodies of respective species were then used and immunoblot signals were visualized by ECL.
LC-MS/MS analysisTotal proteins were collected, separated by 17% SDS Gel, stained with Commassie Blue, and the bands at corresponding molecular weight were excised and subjected to trypsin digestion. The resulting peptides were analyzed by QExactive mass spectrometer coupled to a nano-LC (AdvanceLC, Michrom Inc.) The acquired spectra were analyzed with the SEQUEST HT algorithm.
ImmunoprecipitationHEK293T cells were transfected with different plasmids for 72 h. Cells were lysed in ice-cold lysis buffer (0.3% CHAPS, 10 mM β-glycerol phosphate, 10 mM pyrophosphate, 40 mM HEPES (pH 7.4), 2.5 mM MgCl2 and EDTA-free protease inhibitor). The soluble fractions from cell lysates were immunoprecipitated with primary antibodies against RASON, KRAS, FLAG, NF1, Rho, cdc42, or ARHGAP10 by incubation overnight at 4 °C. Protein A or G beads were then added and incubated for 2 h at room temperate. Immunoprecipitants were washed five times with PBST and subjected to immunoblotting.
RAS activity assayGTP-bound RAS (active Ras) was measured using the C-Raf RAS-binding-domain (RBD) pull-down and detection kit (8821, Cell Signaling Technology) as instructed by the manufacturer. In brief, for the negative control, 500 µL cell lysates were treated with 10 µL 0.5 M EDTA, pH 8.0 (for a final concentration of 10 mM) followed by adding 5 µL of 100 mM GDP (for the final concentration of 1 mM). The mixture was incubated at 30 °C for 15 min with constant agitation and the reaction was stopped with 32 µL of 1 M MgCl2 (for final concentration of 60 mM). All samples (but not including the Input one) were mixed with GST-Raf-RBD and glutathione resin and incubated at 4 °C for 30 min. Bounded proteins were eluted and analyzed by immunoblot.
ImmunofluorescenceCultured cells were fixed with 4% formaldehyde for 10−15 min and then blocked with 3% BSA and 0.1% Triton X-100 in PBS for 20 min at room temperature. Immunostaining was performed using the following primary antibodies: anti-RASON (This study), anti-KRAS (Abcam, Waltham, USA, Ab180772). Nuclei were counterstained with DAPI. Images were taken with an Olympus FV1000 confocal microscope (Olympus, Japan) or Leica Inverted Confocal SP8 (Leica, Germany).
EdU incorporation assayEdu incorporation into proliferating cells were measured using an EdU assay kit (Ribobio, Guangzhou, China, C10310-1) following the manufacturer’s instructions. In brief, PDAC cells were cultured at 3 × 103 cells per well in 96-well plates in triplicate for 36–48 h. Cells were then exposed to 50 μM EdU for 2 h at 37 °C and washed with PBS three times. Next, cells were fixed with 4% formaldehyde for 30 min at room temperature and washed with PBS three times, followed by treatment with 0.5% Triton X-100 for 10 min at room temperature. After three PBS washes, the cells were added with 100 μL of Apollo® reaction cocktail for 30 min and washed with PBS three times. Then 100 μL Hoechst 33342 (5 mg/mL) was used to stain the cells for 30 min followed by three PBS washes. Representative images were taken with an Olympus fluorescence microscope.
Cell proliferation assayThe cell counting kit-8 assay (CCK8, Dojodo, Tabaru, Japan, CK04) was performed to measure cell proliferation. Five hundred cells were seeded into each well of 96-well plates and allowed to grow or be treated with drugs for different amounts of time as indicated by each experiment. Cell numbers were determined following the manufacturer’s protocol and absorbance was measured at 450 nm on a microplate reader (Thermo Fisher, MA, USA).
Colony formation assayFor colony formation assays, 300 cells per well were seeded into 6-well plates and incubated for 2 weeks with complete medium. The colonies were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet and counted under a microscope.
3D anchorage-independent growth assay3D colony formation assay was performed to measure malignant transformation of MEF cells, including Ras-less MEF cell lines and primary KRASG12D MEF cells. Briefly, 1–5 × 103 Rason-KO or control MEF cells were mixed with 0.6% agarose (Sigma, MO, USA, A9414) and seeded into low attachment 96-well plates (Thermo Fisher, MA, USA, 168136) and cultured in DMEM/F12 supplemented with BSA (Sigma, A9576), EGF (Peprotech, Cranbury, USA, GMP100-15), FGF (Prepotech, 100-26) and insulin (Sigma, I3536). Colonies were photographed and counted in 1−2 weeks.
ImmunohistochemistryTumor xenografts or surgical specimen tissue slides were deparaffinized, rehydrated through an alcohol series followed by antigen retrieval with sodium citrate buffer. Tumor sections were blocked with 5% normal goat serum (Vector) with 0.1% Triton X-100 and 3% H2O2 in PBS for 60 min at room temperature and then incubated with anti-RASON (1:100, this study), anti-p-C-RAF (CST, Danvers, USA, 9421, 1:100), anti-p-ERK (CST, 9121, 1:100), anti-p-AKT (CST, 3787 1:100) overnight. Expression levels of those antigens were then detected by HRP-conjugated DAB.
Alcian blue and sirius red stainingAlcian blue and Sirius red staining of mouse pancreas tissues were carried out according to published protocols.36 The expression of mucin was stained with Alcian blue reagents (IHC WORLD, Woodstock, USA, IW3000) according to manufacturer’s instructions. Briefly, pancreas sections were deparaffinized, hydrated, and immersed in Alcian blue solution for 30 min at room temperature. After washing with distilled water, the slides were counterstained in nuclear fast red or eosin. Strongly acidic mucosubstances will be stained blue. The presence of collagen was stained with Sirius red (IHC WORLD, Woodstock, USA, IW3012). In brief, slides were deparaffinized and hydrated and nuclei were stained with Weigert’s hematoxylin for 10 min. Following 10 min of washing in running tap water, the slides were stained in picro-Sirius Red solution containing 0.1% Sirius Red in saturated aqueous solution of picric acid for 1 h. Collagen-rich tissue is stained red on a pale-yellow background.
Protein expression and purificationFor eukaryotic expression, RASON ORF was cloned into the pcDNA3.1 vector and transfected into HEK293T cells using Sinofection Transfection Reagent (Sino biological Inc. Beijing, China, STF02), and cultured with SMM 293-TI medium (Sino biological Inc. Beijing, China, M293TI-1). Shake flask culture condition: 37 °C, 5% CO2, 175 rpm table speed. One week after transfection, the medium was collected and filtered through 0.45 µm filters. The purification process contained the following steps: Step 1, the Ni2+ packed column was washed with ddH2O and equilibrated with binding buffer (50 mM Tris-HCl, 150 mM NaCl, pH 8.0) at a flow rate of 0.75 mL/min. Step 2, samples were then loaded onto the Ni2+ packed column at a flow rate of 0.5 mL/min. Step 3, elution buffer was used to elute unbound proteins and molecules (washed twice with 50 mM imidazole and 100 mM imidazole, respectively). Step 4, RASON protein was obtained highly purified by using elution buffer (300 mM imidazole). Step 5, RASON protein was verified by immunoblotting. Step 6, RASON protein was washed with wash buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl) at a flow rate of 0.75 mL/min and kept in −80°C.
For prokaryotic expression, human RASON protein fused with a C-terminal TEV protease cleavage site and Tamavidin 2 protein was incorporated into the pET-30a vector between NdeI and XhoI endonuclease cleavage sites with a 6× His tag at the C-terminus. The protein was overexpressed in BL21(DE3) bacteria as incubated at 37 °C and induced with 1 mM IPTG. After 4 h of expression, the cells were collected by centrifugation at 4 °C and 4000 rpm for 10 min, and then suspended in the buffer of 50 mM Tris-HCl, pH 8.0, 300 mM NaCl, and 2 mM DTT and lysed by sonication in an ice-water bath. The inclusion bodies were washed twice using the lysis buffer and then dissolved in 8 M urea and loaded onto a nickel column. After removing urea step wisely, TEV protease was added to cleave the recombinant protein at 4 °C overnight on the column. The eluted RASON was further purified by Superdex 75 10/300 GL gel-filtration column (GE Healthcare) and the protein concentration was measured by NanoPhotometer N50.
KRAS coding sequence was synthesized by GenScript (Nanjing, Jiangsu, China) and cloned into the pET-30a (+) vector. KRASG12D and KRASG12V plasmids were generated through point mutation by high-fidelity PCR. 6×His-tagged recombinant human KRASG12D and KRASG12V proteins (residues 1–169) were produced in Escherichia coli (E. coli) BL21(DE3) using published protocols.10 The GAP-related domain (GRD) of NF1 protein (residues 1198–1530) was cloned into the pET-24d vector and expressed in E. coli BL21(DE3) as a recombinant protein with N-terminal 6×His-tag using published protocols.37 Ras-specific nucleotide exchange factor SOScat (residues 564–1049) was cloned into the pET-30a vector and produced in E. coli BL21(DE3) as reported.38 For NMR titration, 15N-labeled KRASG12D protein was produced in E. coli BL21(DE3) which was cultured in M9 medium (15NH4Cl) with 5 g/L glucose, 2 mM MgSO4 and 0.1 mM CaCl2, and purified as mentioned above.
NMR experimentAll NMR experiments were carried out at 298 K on a Bruker AVANCE NEO 800 MHz spectrometer equipped with a 5 mm z-gradient 1H&19F/13C/15N TCI cryogenic probe. The 2D 1H-15N HSQC spectra of 15N-labeled KRASG12D (20 μM) in the absence and presence of unlabeled RASON were obtained to detect the interaction. All NMR spectra were processed and plotted by topspin 4.1.1.
GTP hydrolysis assayThe malachite green assay was used to measure intrinsic and extrinsic GTP hydrolysis rates of KRASG12D and KRASG12V by detecting the production of inorganic phosphates.39 Briefly, KRASG12D or KRASG12V protein was first loaded with GTP (Sigma, MO, USA, 11140957001) for 2 h at 4 °C. For intrinsic hydrolysis, 7.5 µL GTP-loaded KRAS protein in reaction buffer (10 mM Tris, pH 7.6, 150 mM NaCl, 2 mM MgCl2, 0.05% Tween-20) was added to wells of clear flat bottom 384-well plates (JET BIOFIL, Guangzhou, China, TCP011384). Then 0.5 µL mixture including RASON and SOS1 was added to start the reaction and the reaction mixture was allowed to incubate at 37 °C from 0−10 h by starting the 10 h reaction first and then subsequent reactions at each time point. Finally, 2 µL EDTA was added to each well to stop reaction. The final reaction volume in each well is 30 µL. The concentrations of each component in the final mixture were as follows: KRAS (4.0 µM), SOS1 (1.0 µM), GTP (100 µM), EDTA (100 µM), RASON (0.0 µM, 0.1 µM, 0.3 µM, 1.0 µM, 3.0 µM, 10.0 µM). The optical absorption was measured by a Spectramax M4 plate reader at 650 nm (Molecular Device, San Jose, USA) after adding 20 µL malachite green to each well. Samples were measured in duplicates for each experiment.
For extrinsic hydrolysis, the experiment was carried out similarly as described for the intrinsic hydrolysis assay using the same plate set-up and plate reader. In brief, 7.4 µL GTP-loaded KRAS protein in reaction buffer was added to each well. Then 0.6 µL extrinsic mixture including ingredients of intrinsic mixture plus NF1 (RAS-GAP) was added to corresponding wells followed by incubation for specified durations at 37 °C. For extrinsic hydrolysis of KRAS, the duration of incubation was set as follows: 0 s, 30 s, 60 s, 120 s, 300 s, 600 s, 1800 s, 3600 s, respectively. Finally, 2 µL EDTA was added to stop reaction. The concentrations of each component in the final mixture were the same as the intrinsic hydrolysis assay except for NF1 (1.0 µM). To eliminate contaminating background phosphate signal, we included a control group with all ingredients except KRAS for every corresponding experiment.
Nucleotide exchange assayNucleotide exchange assay for KRASG12D and KRASG12V proteins were carried out according to published protocols.14 Briefly, KRAS proteins (10 µM) were loaded with 200 µM mant-GDP (Sigma, MO, USA, 69244) in the presence of 2.5 mM EDTA (Sigma, E5134). After incubation for 1 h at room temperature in dark, 10 mM MgCl2 was added to terminate the reaction. The proteins were desalted by NAP-5 columns (GE, Chicago, USA, 17085302). 10 µL of each desalted protein was added to low-volume black bottom 384-well plates (Corning, NY, USA, 4514) followed by 5 μL GMPPNP (sigma, G0635). To initiate the nucleotide exchange reaction, 5 μL of different concentrations of RASON (0.0 μM, 0.3 μM, 1.0 μM, 3.0 μM, 10.0 μM final) with (extrinsic) or without (intrinsic) SOS (1 μM final), EDTA (5 mM final), or reaction buffer was added and fluorescence was monitored on a Spectramax M4 plate reader (355 nm excitation, 448 nm emission) for 1 h at 90-s intervals.
SPRA Biacore S200 instrument (GE Healthcare, Chicago, USA) was used to detect protein–protein interactions using a direct binding assay format. Prior to activation, the CM5 chip surface was preconditioned using two 50 μL injections each of 10 mM HCl, 50 mM NaOH, 0.1% SDS, and 0.085% H3PO4 at a flow rate of 100 μL/min. RASON was immobilized on the sensor surface using standard amine coupling. This was achieved by activating the sensor surface using 7 min injections of a mixture of 11.5 mg/mL N-hydroxysuccinimide with 75 mg/mL 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride. Protein immobilization was accomplished using a 10 min injection of protein (30 μg/mL) in 10 mM NaAc, pH 5.0 buffer. Remaining reactive esters were blocked using a 7 min injection of 1 M ethanolamine, pH 8.5, at a flow rate of 10 μL/min. Reference flow cells were prepared without the protein. Denaturation was achieved by a 30 s injection of 10 mM HCl at a flow rate of 30 μL/min. All binding measurements were performed in 0.01 M HEPES, pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% surfactant P20, at a flow rate of 30 μL/min. KRASG12D or KRASG12V was injected over the immobilized RASON protein and reference surface with at least 30 s association and dissociation time. Surface regeneration was achieved by dissociation for a time period allowing the response to return to baseline. Control injections of different concentrations of KRASG12D and KRASG12V were used to allow monitoring of the functionality of the protein surface. SPR equilibrium binding data, consisting of Req values from several concentration series, were analyzed by fitting a simple 1:1 binding to yield Rmax and Kd values using Biacore S200 Evaluation Software.
Quantification of KRAS, NF1 and RASON protein levels in cellsLC-MS was used to quantify the protein levels of KRAS, NF1, and RASON in AsPC-1 and PANC-1 pancreatic cancer cell lines. Purified KRAS, NF1, and RASON proteins were first serial diluted, digested, and analyzed by NanoLC/Triple TOF 5600 (AB SCIEX, Framingham, USA) to generate a standard curve for each protein. Total proteins from cell lysates were separated by 15% SDS-PAGE, stained with Commassie Blue, and the bands at corresponding molecular weight were excised and subjected to trypsin digestion. The resulting peptides were analyzed by LC-MS. The acquired data were analyzed with the Peakviewer 2.0 and Proteinpilot software. Intracellular levels of each protein were then determined by detection and quantification of the respective signature peptides according to the standard curve.
Mice and animal housingAthymic (Ncr nu/nu) mice at 6–8 weeks of age were purchased from GemPharmatech Co, Ltd (Nanjing, China). All mice were housed in a SPF facility (5 mice per cage) under a 12-h light-dark cycle with free access to food and water. All experiments using animals were conducted under the Institutional Animal Care and Use Committee (IACUC)-approved protocols at Nanjing University (IACUC-2101008) or Sun Yat-sen University (2021878) in accordance with NIH and institutional guidelines.
Xenograft studiesIn vivo PDAC xenograft growth studiesMice were randomly assigned to experimental groups for all the experiments. For PDAC cell line xenografts, 1 × 107 BXPC-3 cells or 5 × 106 AsPC-1 and PANC-1 cells in 100 μL culture media/Matrigel (4:1) were injected subcutaneously.
Subcutaneous tumor formation of MEF cellsRas-less MEF cells (1.5–2 × 106) with different genetic manipulations were injected subcutaneously into the dorsal flanks of 6-week-old male athymic nude mice, with each mouse bearing one control tumor and one Rason-KO tumor. Mice were euthanized when the larger tumor reached 1000 mm3. The tumors were then excised and measured with a caliper in two dimensions.
In vivo xenograft treatment studiesIn the treatment studies, mice bearing AsPC-1 xenografts were randomly assigned to five groups: control, cetuximab (10 mg/kg, twice a week; MCE, Monmouth Junction, USA, HY-P9905), Blank (AAV9) + cetuximab, shRASON (AAV9) (1 × 1013 vg/mL, 100 μL peritumoral injection at day 7 and 21; Vigene Bioscience, Jinan, China), and cetuximab + shRASON (AAV9). Tumors were measured with digital caliper twice a week, and tumor volumes were determined with the formula: (length × width2)/2 and plotted as means ± SEM.
CRISPR-mediated generation of Rason −/− miceThe Rason−/−mouse strain was established using CRISPR-mediated deletion of the ORF of mouse Rason. The target sequences of gRNA were: gRNA1: CGCGGATTGTGTGTTTGCGT; gRNA2: GGGTTGGGGTTCCCCTAACG. The Rason-KO allele was bred into C57BL/6 background; and animals were maintained and crossed using standard procedures (Cyagen Biosciences, Guangzhou, China). The following primers were used to identify the homozygous KO mice (forward: 5′-CAACTCCCAGGATACCATTCGGC-3′; reverse: 5′-GAGCCCAGAACAGCCGCTGAC-3′).
Genetic mouse models for pancreatic tumorigenesisThe LSL-KrasG12D; Pdx1Cre (KC) genetic mouse strain described previously40,41 was provided by Shanghai Model Organisms Center, Inc (Shanghai, China) and crossed with Rason−/− mice to generate LSL-KrasG12D; Pdx1Cre; Rason−/− (KCR) mice.
The LSL-KrasG12D; Trp53R172H/+; Pdx1Cre; Rasonmut/mut (KPCR) mouse strain was generated via in vitro fertilization (IVF) by GemPharmatech Co, Ltd (Nanjing, China). In brief, two start codons, the first and the fifteenth ATG, of Rason were mutated to stop codon TAA by CRISPR in LSL-KrasG12D; Trp53R172H/+ (KP) mice during the first IVF to obtain LSL-KrasG12D; Trp53R172H/+; Rasonmut/mut (F0) mice, which were used for the second IVF with Pdx1Cre mice to generate F1 mice. The third IVF was carried out among different genotypes of F2 mice, but only four target mice were obtained from 741 F3 offspring. The fourth IVF was carried out using F3 mice with different genotypes to obtain enough LSL-KrasG12D; Trp53R172H/+; Pdx1Cre; Rasonmut/mut (KPCR) mice. A detailed scheme was also provided in Supplementary information, Fig. S10.
Genetic mouse model for lung tumorigenesisThe LSL-KrasG12D; Trp53R172H/+ (KP) mice were provided by GemPharmatech, Inc (Nanjing, China). A lentivirus (OBiO Technology, Shanghai, China) containing Cre, two mouse Rason sgRNAs (AAAGGCGCAGAGCTGTACG, ACCCACGCGGGGCCGCGGCG), and spCas9 was intratracheally administered at 105 infectious particles to initiate autochthonous lung cancer.42 Mice were anaesthetized to evaluate tumor growth by microcomputed tomography (micro-CT) scanning or euthanized to confirm lung adenocarcinoma formation by histological analysis.
Micro-CT scanningMicro-CT scans were performed using Hiscan XM Micro CT (Suzhou Hiscan Information Technology, Suzhou, China). The X-Ray tube settings were 60 kV and 133 uA and images were acquired at 50 µm resolution. A 0.5° rotation step through a 360° angular range with 50 ms exposure per step was used. The images were reconstructed with Hiscan Reconstruct software (Version 3.0) and analyzed with Hiscan Analyzer software (Version 3.0).
Human pancreatic tumor organoid culture and drug treatmentHuman PDAC PDOs were established according to the protocol established by the Tuveson group.43 In brief, freshly collected tumor tissues from PDAC patients undergoing surgery were minced and digested with 5 mg/mL collagenase II (Thermo Fisher, MA, USA, 17101015) and 10 μg/mL DNase1 (Sigma, MO, USA, D5025) in advanced DMEM/F12 supplemented with HEPES (Thermo Fisher, MA, USA, 15630080), Glutamax (Thermo Fisher, 35050061), and Penicillin/Streptomycin (Thermo Fisher, 15140122) at 37 °C for 20 min. Cells were then seeded in growth factor reduced Matrigel (Corning, NY, USA, 356231) and cultured in human complete culture medium [Advanced DMEM/F12 medium supplemented with HEPES (Thermo Fisher, 15630080), Glutamax (Thermo Fisher, 35050061), penicillin/streptomycin (Thermo Fisher, 15140122), B27 (Thermo Fisher, 17504044), Primocin (100 µg/mL, InvivoGen, Toulouse, France, ant-pm-1), N-acetyl-L-cysteine (1.25 mM, Sigma, A9165), Wnt3a-conditioned medium (50% v/v), RSPO1-conditioned medium (10% v/v), recombinant mNoggin protein (0.1 μg/mL, Peprotech, Cranbury, USA, 250-38), human epidermal growth factor (hEGF, 50 ng/mL, Peprotech, GMP100-15), hGastrin (10 nM, R&D, 3006), fibroblast growth factor 10 (FGF10, 100 ng/mL, Prepotech, 100-26), Nicotinamide (10 mM, Sigma, N0636), PEG2 (1 μM, R&D, 2296/10), Y-27632 (10.5 μM, Sigma, Y0503) and A83-01 (0.5 μM, R&D, 2939)].
For tetracycline-inducible KD of RASON, the organoids were first digested into single cells with TrypLE (Thermo Fisher, MA, USA, 12605010) and transfected using lentivirus carrying the shRNA construct. Three days later, puromycin was added for selection. When needed, 1 µg/mL tetracycline (MCE, Monmouth Junction, USA, HY-A0107) was added to induce RASON shRNA (GGGAAGGACTGATCCACATTC) expression.
For combination therapy with cetuximab and RASON shRNA, organoids were seeded into 48-well plates and recovered for seven days before cetuximab treatment. shRASON was activated by tetracycline three days before cetuximab treatment. Organoids were treated with cetuximab (10 µg/mL, MCE, Monmouth Junction, USA, HY-P9905) with or without shRASON for 9 days and imaged every 3 days. At the end of experiment, CellTiter-Glo 3D Reagent (Promega, Madison, USA, G9683) was used to measure cell viability according to the manufacturer’s instructions.
Statistical analysisStatistical analyses were performed using GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA). The statistical tests performed were paired student’s t-test, multiple t-test, one-way ANOVA, two-way ANOVA, Wilcoxon test, or log-rank analysis. Sample sizes (n) are indicated in the figure legends. Significance was set as *P < 0.05, **P < 0.01, ***P < 0.005 or ****P < 0.0001 for all data.
Materials availabilityAll stable and unique reagents generated in this study are available upon reasonable request.
留言 (0)