
記住我


The bottleneck in the structural analysis of human Cav3.2 arises from the low yield of recombinant protein expression. To enhance protein production, various constructs were explored. The I-II loop plays an inhibitory role in Cav3.2 expression and function.48,49 We thereby introduced several internal truncations of different fragments to this region. Eventually, a variant with the deletion of residues 493-772 resulted in an elevated expression level and decent solution behavior (Supplementary information, Fig. S1). This variant was named Cav3.2EM, as it was used for cryogenic electron microscopy (cryo-EM) imaging.
The biophysical properties of Cav3.2EM were verified through whole-cell patch-clamp recordings. Consistent with its improved protein expression, Cav3.2EM exhibited an increased conductance compared with the wild-type (WT) channel. Additionally, both the activation and steady-state inactivation curves of Cav3.2EM demonstrated a slight leftward shift in comparison to WT (Fig. 1a; Supplementary information, Fig. S2 and Table S1).
Fig. 1: Cryo-EM structural analysis of human Cav3.2.
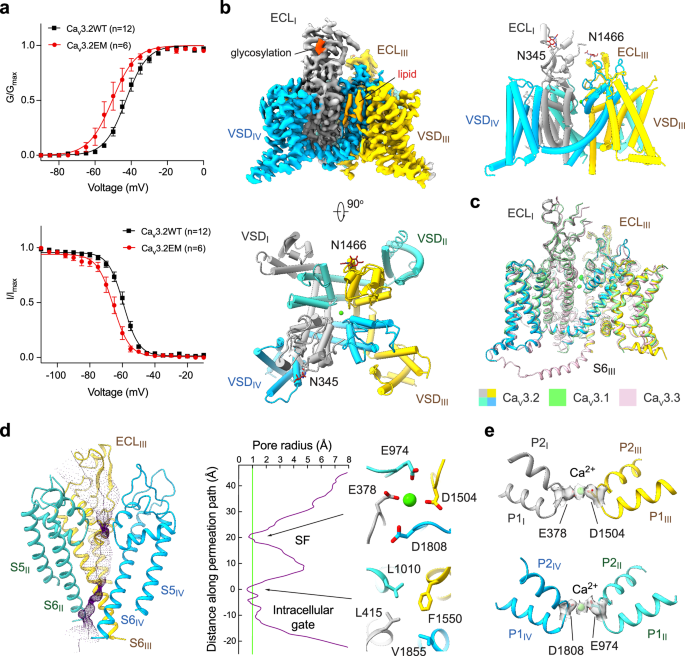
a Electrophysiological characterizations of full-length human Cav3.2WT (black) and truncated Cav3.2EM (red) in HEK293T cells. Voltage-dependent activation and steady-state inactivation curves are presented in the upper and lower panels, respectively. n values indicate the number of independent cells; data are presented as mean ± SEM. Please refer to Materials and Methods and Supplementary information for details. b Cryo-EM reconstruction of human Cav3.2EM. The EM map is color-coded for the four repeats (upper left), and the sugar moieties and lipids are colored bright and pale orange, respectively. The same color scheme is applied throughout the manuscript. The overall structure of human Cav3.2 is shown in a side view (upper right) and top view (bottom). c Structural comparison of the three T-type Cav channels: Cav3.1 (PDB: 6KZO, light green), Cav3.2 (domain colored), and Cav3.3 (PDB: 7WLI, pink). d The ion-conducting path of Cav3.2 is calculated in HOLE70 and illustrated with purple dots (left). The calculated pore radii along the permeation path are depicted as a purple line (middle). Two constriction sites, the SF enclosed by the EEDD motif and the intracellular gate, are shown on the right, in extracellular views. e Ca2+ coordination in the SF. Densities for the SF, prepared in ChimeraX, are contoured at the level of 4 σ, and the potential Ca2+ ion is shown as a green sphere.
Following the functional validation of Cav3.2EM, we performed standard cryo-EM single particle analysis and obtained a three-dimensional (3D) EM reconstruction at an overall resolution of 3.0 Å. This structure is henceforth referred to as Cav3.2Apo. Two glycan chains were observed, attaching to Asn345 and Asn1466 in the extracellular loops in repeats I and III (ECLI and ECLIII) via N-link (Fig. 1b; Supplementary information, Figs. S3, S4 and Table S2). Previous studies have also suggested the importance of these N-glycosylation sites in regulating the functional expression of Cav3.2.50,51,52
The overall structure of Cav3.2Apo displays an inactivated state nearly identical to that observed for Cav3.1 and Cav3.3,44,47 with root-mean-square deviations (RMSDs) of 0.58 Å over 847 Cα atoms and 1.16 Å over 948 Cα atoms, respectively (Fig. 1c). All four voltage-sensing domains (VSDs) adopt the depolarized or “up” conformation, and the intracellular gate is tightly twisted. The closed intracellular gate comprises three layers of hydrophobic residues. The first assembly site beneath the central cavity includes conserved residues Leu415, Leu1010, Phe1550, and Val1855, a common feature observed in the structures of all three members of Cav3 channels (Fig. 1d). A spherical density, likely corresponding to a Ca2+ ion, is surrounded by four residues, Glu378, Glu974, Asp1504, and Asp1808 (the EEDD motif) in the selectivity filter (SF) (Fig. 1e).
Structural determination of Cav3.2 with different antagonistsNext, we set out to determine the structures of Cav3.2 in complex with representative T-type channel-selective antagonists. To validate the action of these compounds on both WT Cav3.2 channel and Cav3.2EM variant, we conducted whole-cell patch-clamp recordings in HEK293T cells (Supplementary information, Fig. S5 and Tables S3, S4). The characterizations indicated that Cav3.2EM exhibited comparable potency across all tested compounds compared to the WT channel. Each antagonist was individually incubated with purified Cav3.2 protein at a final concentration at least 10-fold higher than its IC50 value before cryo-sample preparation. Following similar protocols for cryo-EM data acquisition and analysis, we successfully resolved the structures of Cav3.2 in complex with four compounds, ACT-709478, TTA-A2, TTA-P2, and ML218, with overall resolutions ranging from 2.8 Å to 3.2 Å. For simplicity, we will refer to these structures as Cav3.2-ACT/TA/TP/ML, with “ACT” representing ACT-709478, “TA” for TTA-A2, “TP” for TTA-P2, and “ML” for ML218 (Supplementary information, Fig. S6 and Table S2).
All four complex structures resemble the apo form, with the compounds each accommodated within the pore domain (PD) (Supplementary information, Fig. S6q). The four compounds can be further categorized into two groups: ACT-709478 and TTA-A2 insert through the IV-I fenestration, while TTA-P2 and ML218 dock on the II-III fenestration. Apart from the fenestration binding site, the other end of these elongated compounds is nestled within the central cavity. The fenestration-accommodating binding poses immediately suggest the molecular basis for their state-dependent pore-blocking mechanism. In the following text, we will illustrate their binding details, which will facilitate future drug design and optimization.
Coordination of ACT-709478 and TTA-A2Both ACT-709478 and TTA-A2 were well resolved. The distinct structural features within the densities, like the trifluoromethyl group in ACT-709478 and the methyl group in TTA-A2, enabled reliable model building of these small-molecule compounds (Supplementary information, Fig. S6).
The head groups, characterized by the cyclopropylphenyl group in TTA-A2 or the trifluoromethyl cyclopropylphenyl moiety in ACT-709478, reside in the center of the cavity, which we named site C in Nav channels53,54 (Fig. 2a, b). These groups are coordinated similarly by several hydrophobic residues from the S6 tetrahelical bundles, including Phe408 and Asn412 on S6I, Phe1007 and Leu1010 on S6II, Val1546, Leu1547, and Phe1550 on S6III, and Gln1848, Leu1851, and Val1852 on S6IV (Fig. 2c, d).
Fig. 2: Molecular basis for Cav3.2 inhibition by ACT-709478 or TTA-A2.
a Chemical structures of ACT-709478 and TTA-A2. The two compounds share a common core structure, featuring a cyclopropylphenyl head (highlighted in red), an aromatic tail (highlighted in green), and an amide linker. b Structural basis for pore block by ACT-709478 or TTA-A2. Both molecules traverse the central cavity, with one end inserting into the IV-I fenestration. The two structures, named Cav3.2-ACT and Cav3.2-TA, are superimposed relative to the PD. ACT-709478 and TTA-A2 are shown as brown and black sticks, respectively, and only the PD of Cav3.2-ACT is shown. c Detailed coordination of ACT-709478 and TTA-A2. The aromatic tail groups of ACT-709478 and TTA-A2, which vary in details, can both be accommodated in the IV-I fenestration. Potential H-bonds are highlighted with red dashed lines. d Schematic representation of residues constituting the binding site for ACT-709478 (upper) and TTA-A2 (lower). Residues within a 4 Å cutoff distance to the ligand are shown, with the binding pocket and potential H-bonds indicated by gray dashed contour and red dashed lines, respectively.
Both tail groups, despite their distinct chemical structures, wedge into the IV-I fenestration, indicating the fenestration’s versatile adaptability to diverse molecules. Per our recently proposed nomenclature system for the druggable sites on Nav channels,53 this site will be described as site F4. The tail groups in the IV-I fenestration are surrounded by hydrophobic residues, Leu377, Ile403, Ser407, and Phe408 from repeat I, as well as Phe1756, Phe1802, Ser1805, and Thr1806 from repeat IV. In the case of ACT-709478, the nitrogen within the cyanopyridine ring is further stabilized through a hydrogen bond (H-bond) with the hydroxyl group from Ser1805. Furthermore, both internal amide linkages are H-bonded with Asn412 and Gln1848, contributing to the stability of these unique binding poses (Fig. 2c, d).
An α-to-π transition of S6II in the presence of TTA-A2ACT-709478 and TTA-A2 share a similar pharmacophore featuring a cyclopropylphenyl head, an aromatic tail, and an amide linkage. However, there are local structural variations in the presence of these two compounds. While the conformation of Cav3.2-ACT is nearly identical to that of the apo channel, structural rearrangements occur in the presence of TTA-A2, as exemplified by an α-to-π transition in the middle of the S6II segment (Fig. 3a).
Fig. 3: Local structural shifts in the presence of TTA-A2.
a An α-to-π transition observed in the S6II segment upon TTA-A2 binding. Left: Red arrows indicate the structural differences between Cav3.2-ACT (gray) and Cav3.2-TA (domain colored). An enlarged view that highlights the change of S6II is shown in the inset. Right: Rotation of the bottom half of the S6II helix in the presence of TTA-A2, but not ACT-709478. b Closure of the I-II fenestration in Cav3.2-TA. Corresponding surface views of the four sides of the PD are presented for Cav3.2-ACT (upper) and Cav3.2-TA (lower).
A close structural comparison of Cav3.2-ACT/TA reveals that the conformational deviation is caused by the minor difference in the head group. The only divergence of the head group of the two molecules pertains to the presence of an additional trifluoromethyl group in ACT-709478. This protrusion would clash with Phe1007 in a π-helical configuration but align well with the α-helix. Yet, Phe1007 provides a favorable environment for accommodating the smaller cyclopropylphenyl group in TTA-A2, explaining the π form of S6II in Cav3.2-TA (Fig. 3a). As a result of the minor rotation of Phe1007, the I-II fenestration, present in the apo channel and Cav3.2-ACT, diminishes in Cav3.2-TA, and the gating residue on S6II shifts from Leu1010 to Val1011 in Cav3.2-TA, with the intracellular gate remaining closed (Figs. 1d, 3b; Supplementary information, Fig. S7).
TTA-P2 and ML218 bind through the II-III fenestrationTTA-P2 and ML218 also share a similar chemical structure, characterized by a 3,5-dichlorobenzamide head and an aliphatic tail (Fig. 4a). They also display a similar binding paradigm, with the head adhering to the II-III fenestration (Site F2), and the tail projecting into the cavity (site C) (Fig. 4b). Despite an ~30° deviation of the binding poses for the two head groups, the accommodation site within the fenestration is similar. The environment is primarily hydrophobic, enclosed by residues from S5II, P1II, S6II, and S6III segments, including Leu922 on S5II, Leu971 on P1II, Asn1003 and Phe1007 on S6II, Lys1503 in the P-loop, and Leu1539, Ser1543, Leu1547, and Phe1550 on S6III (Fig. 4c, d).
Fig. 4: Specific inhibition of Cav3.2 by ML218 or TTA-P2.
a Chemical structures of ML218 and TTA-P2. b The two molecules exhibit similar binding poses. Structures of Cav3.2-ML (with ML218) and Cav3.2-TP (with TTA-P2) are superimposed relative to the PD. Only the PD structure of Cav3.2-ML is shown as semi-transparent cartoon. c The binding poses of ML218 and TTA-P2 deviate with an ~30° rotation of the 3,5-dichlorobenzamide head within the II-III fenestration. Detailed coordination of ML218 and TTA-P2 in the II-III fenestration are presented in the middle and right panels, respectively. d Schematic representation of residues constituting the binding site for ML218 or TTA-P2 within a 4-Å cutoff distance. The binding pocket and potential H-bonds are indicated by a gray dashed contour and red dashed lines, respectively. e Coordination of the aliphatic tail of the two compounds in the central cavity of Cav3.2. f Both structures adopt a π configuration in the middle of the S6II segment.
The aliphatic tail of both TTA-P2 and ML218 reclines across the central cavity, directly obstructing ion permeation. The cavity site is formed by Leu415 on S1I, Phe1007 on S6II, and Ser1543, Leu1547, and Phe1550 on S6III (Fig. 4d, e) Similar to the conformation observed in the presence of TTA-A2, S6II adopts a π-helix conformation upon binding to TTA-P2 or ML218. On one end, the 3,5-dichlorobenzamide head group would encounter spatial repulsion with Leu1006 in an α-helix conformation; on the other end, the aliphatic tail could be further stabilized by Phe1007 through a π-H interaction, thus favoring the π-helix form (Fig. 4f).
Stabilization of antagonist binding by an endogenous lipidExamination of the 3D EM maps for all the structures, including that of the apo channel, identifies a well-resolved density in the cavity, which can be best fitted with a phosphatidylethanolamine (PE) molecule (Fig. 5a, b; Supplementary information, Fig. S8). The two hydrophobic tails penetrate the III-IV fenestration, and the polar head inserts into the central cavity, directly contributing to drug coordination. Similar paradigm has been observed in drug-bound structures of Cav1.1, Cav3.1, and Cav3.3.22,44,47
Fig. 5: Stabilization of antagonist binding by endogenous lipids.
a A conserved endogenous lipid stabilizes antagonist association within the central cavity. The EM densities for the lipid and antagonist are contoured at the similar level of 4.5–5 σ. The antagonists show more diverse docking poses in the absence of the endogenous lipid. Predicted binding poses in the absence of the lipid are represented as semi-transparent sticks, while the structure-determined binding pose is shown as solid sticks. b Identical binding pose of the lipid in all blocker-bound Cav3.2 structures. Only the pore-forming helices of Cav3.2-TA are shown in an extracellular view of the superimposed structures of the antagonist-bound pore cavity. c Less favored binding of antagonists to Cav3.2 in the absence of the lipid. Predicted binding poses in the absence of the endogenous lipid exhibit larger variability, as indicated by the increased RMSD values. The average binding free energy (ΔGbinding) was calculated using Prime-MM/GBSA.
To investigate the role of the lipid in drug binding, we performed molecular docking simulations with or without the lipid. The docking poses in the presence of the lipid align well with the experimental structures, with RMSD values of the predicted binding poses < 3.0 Å compared to the corresponding experimental structures. In contrast, in the absence of the lipid, the antagonists display more diverse docking poses, resulting in a broader range of RMSD and ΔGbinding values (Fig. 5a, c). Among these, flipped poses are even more favored for ACT-709478 and TTA-A2 in the absence of the lipid (Fig. 5a). Our computational analyses support the role of the lipid in stabilizing the accommodation of the antagonists in the cavity, yet the physiological relevance and the identity and specificity of the endogenous lipids that may facilitate drug binding awaits further characterizations.
Structural basis for ligand selectivity on T-type Cav channelsThe four structures presented here, along with our previously reported Cav3.1-Z944 complex structure,44 offer important insights into T type-specific inhibition by these selective antagonists. Sequence alignment reveals that several residues involved in ligand binding, including Leu377, Gln973, Phe1007, Leu1010, Val1011, Lys1503, Leu1539, Leu1540, Ser1543, Val1546, Leu1547, Phe1556, Ser1805, Gln1848, Leu1851, Val1852, and Val1855, most of which are positioned on the P-loops and the S6 tetrahelical bundle, vary from those in the HVA Cav channels (Fig. 6a).
Fig. 6: Structural basis for antagonist selectivity on T-type Cav channels.
a Structure-guided sequence analysis to identify the determinants for antagonist selectivity. Sequence comparison of human Cav channels for the antagonist-binding residues is shown. The dashes represent residues in other subtypes that are identical to the corresponding ones in Cav3.2. b Functional validation of residues critical to TTA-A2 selectivity. Several residues in Cav3.2 were mutated to corresponding ones in the HVA channels. The responses of these mutants to TTA-A2 were examined through whole-cell patch-clamp recordings. c A magnified view of the coordination of TTA-P2 and ML218 in the superimposed structures of Cav3.2-TP and Cav3.2-ML. d F1007L confers Cav3.2 resistance to ML218, but not TTA-P2. Experimental details are provided in Materials and Methods and Supplementary information, Figs. S9–S11, and Tables S3 and S4. e Potential molecular basis for the distinct responses to TTA-P2 and ML218 by Cav3.2-F1007L. The additional fluorine atom in the piperidine ring of TTA-P2 may interfere with the essential π-H interaction between Phe1007 and the piperidine ring, potentially resulting in lower potency on Cav3.2, which is largely unaffected by the Leu substitution.
To identify the residues that underlie the subtype-specific sensitivity to these inhibitors, we started with in silico molecular docking and binding free energy calculation for Cav3.2 mutants each with a single locus substituted with the corresponding residue from the HVA channels (Supplementary information, Fig. S9a). The computational analysis suggests that many mutations would have distinct impact on different inhibitors. For example, L377M, Q1848A/S, and L1851V/I/M resulted in decreased binding energy for ACT-709478 and TTA-A2, but not TTA-P2 or ML218. On the other hand, F1007L reduces the affinity to TTA-A2 and ML218, but has little effect on ACT-709478 and TTA-P2 (Supplementary information, Fig. S9).
Based on these clues, we generated a number of corresponding Cav3.2 mutants and characterized their responses to the drugs using whole-cell patch-clamp recordings in HEK293T cells. Several mutations indeed attenuate the potency of the drugs, as exemplified by L377M, F1007L, Q1848A, L1851M and L1851I for TTA-A2, and F1007L for ML218. Consistent with the computational results, F1007L shows little effect on the potency of TTA-P2, which shares a similar binding pose to ML218 (Fig. 6b–d; Supplementary information, Figs. S9–S11 and Tables S3, S4).
The difference in F1007L’s responses to ML218 and TTA-P2 may be attributed to the additional fluorine atom in the piperidine ring of TTA-P2, which interferes with the π-H interaction between Phe1007 and the piperidine ring, leading to an ~30° deviation of the binding poses for the 3,5-dichlorobenzamide heads. Therefore, the potency of TTA-P2 on Cav3.2, regardless of the F1007L mutation, is lower than that of ML218 (Fig. 6c–e; Supplementary information, Fig. S9, Table S4). As there is no fluorine atom in the piperidine group of Z944, an analog of ML218 and TTA-P2, we also performed similar analysis on the complex structure of Z944-bound Cav3.1. Its overall binding pose in Cav3.1 is closer to that of ML218 in Cav3.2 than TTA-P2. Replacing the allelic Phe956 with Leu in Cav3.1 significantly reduces the potency of Z944 on Cav3.1 in a similar manner to that of ML218 on Cav3.2,44 thereby confirming the distinct sensitivity of Phe1007 in Cav3.2, or Phe956 in Cav3.1, in ligand recognition (Supplementary information, Figs. S9–S11 and Table S4).
It is noted that K1503F or K1503G, which only slightly affects the binding energy, does not show a significant impact on the potency of ML218 on Cav3.2 (Fig. 6d). A re-examination of the Z944 response by the corresponding Cav3.1 mutants, K1462F or K1462G, reveals some technical issues in the previous electrophysiological characterizations, which are summarized in the legend of Supplementary information, Table S4. Using a corrected protocol, we show that K1462F or K1462G does not reduce Cav3.1’s sensitivity to Z944 either (Supplementary information, Table S4).44
Taken together, our structural, computational, and functional analyses reveal the complexity underlying the determinants for the subtype specificity by selective inhibitors, and suggest that targeting these critical residues could be a viable strategy for designing selective T-type Cav channel blockers.
留言 (0)