
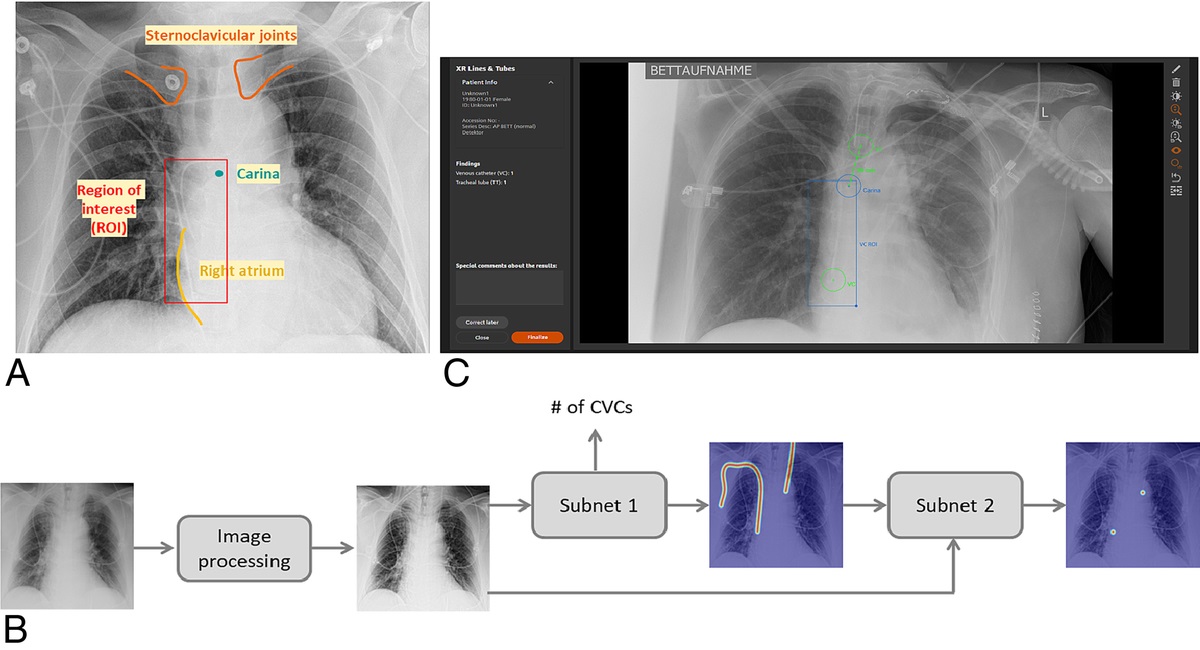
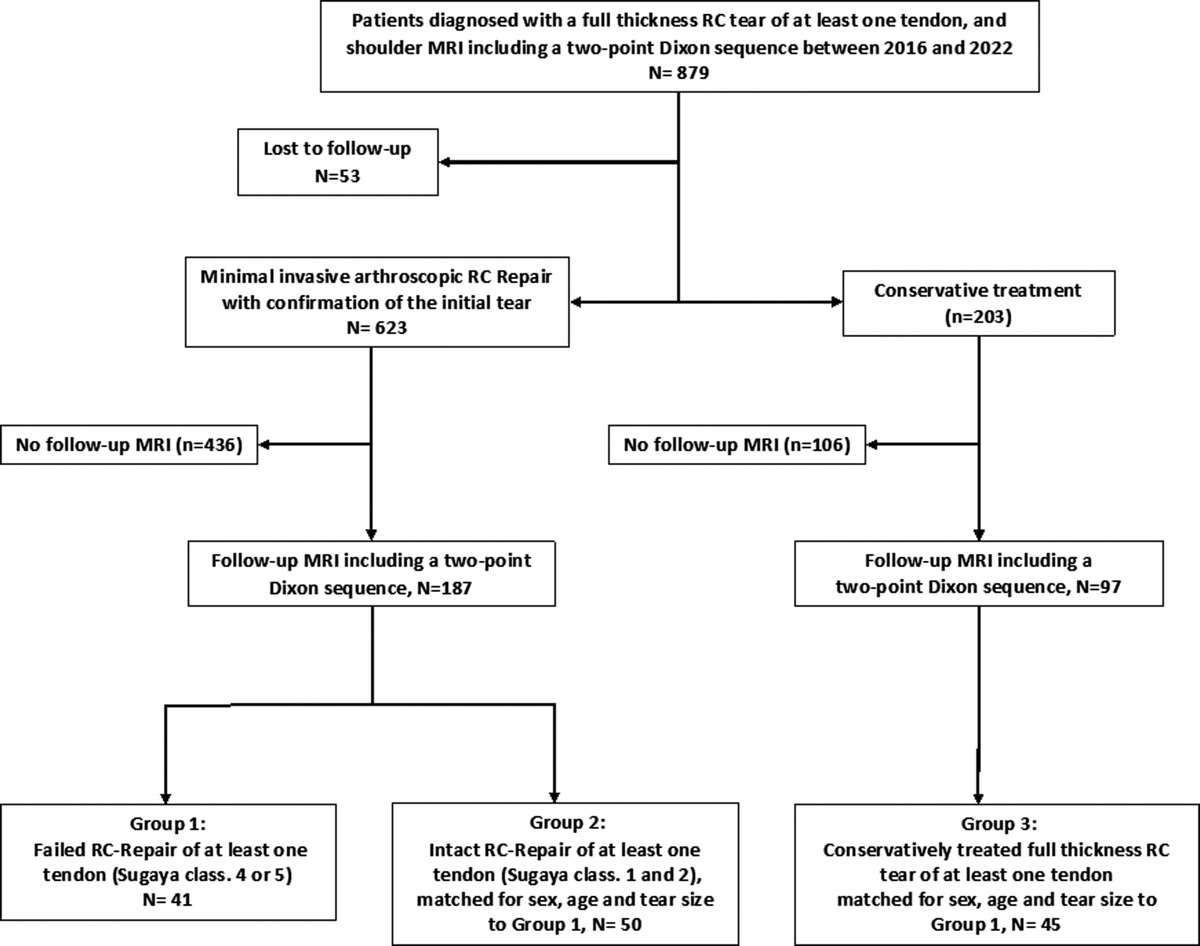
記住我

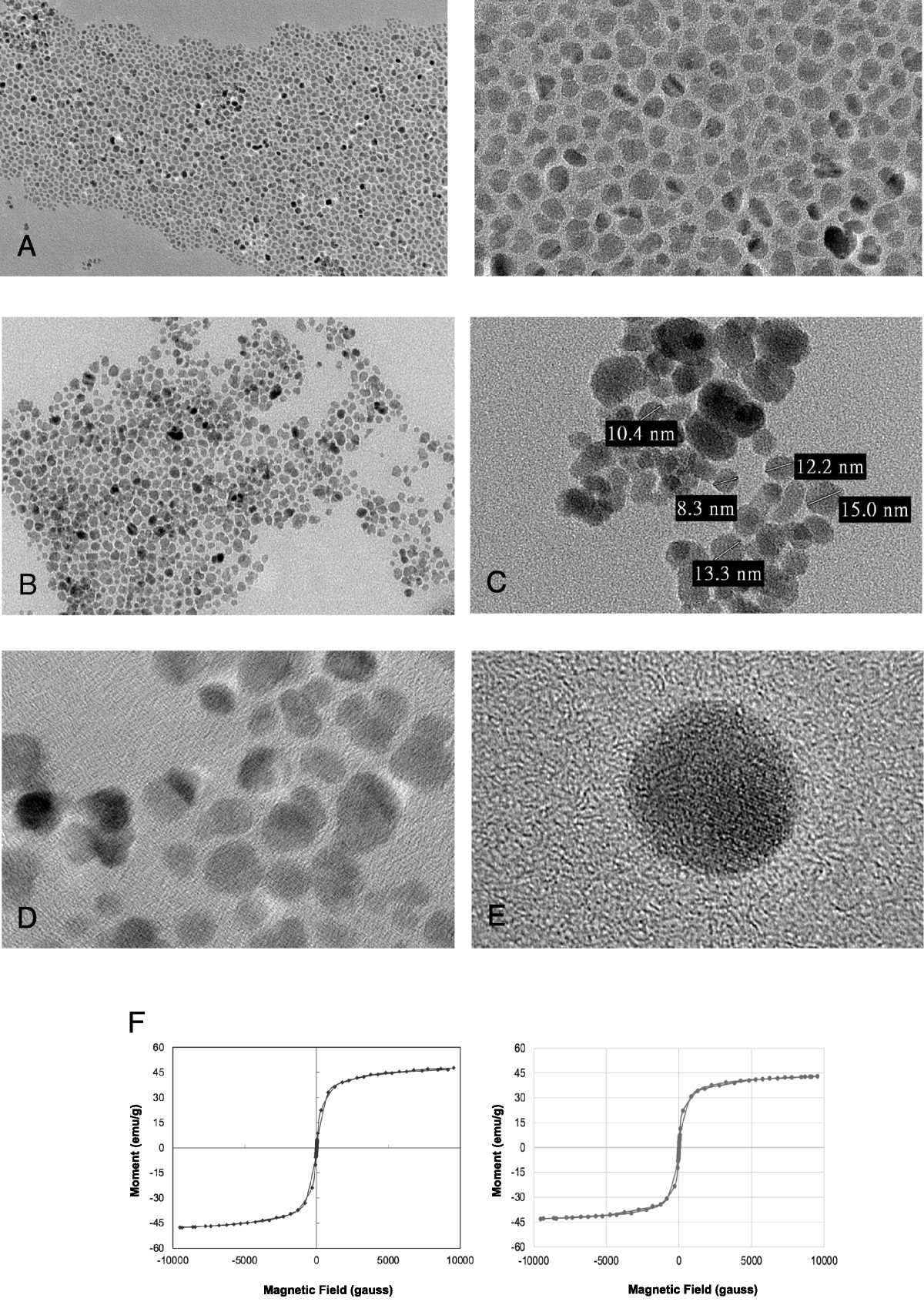



The Gd(III)-based contrast agents (GBCAs) commonly used in contrast-enhanced magnetic resonance imaging (MRI) pass through compromised blood-brain barriers, preferentially accumulating in and enhancing T1 MRI signal in growing brain tumors.1 The lack of a blood-tissue barrier in non–central nervous system (CNS) tissue has made the use of GBCA in the body more challenging. Dynamic contrast-enhanced MRI with GBCAs takes advantage of differences in tumor and tissue vascular permeability to characterize tumors in the body.2 It is thus an important component of multiparametric MRI (mpMRI) for diagnostic imaging and surgery planning of prostate cancer.3,4 Despite some specific clinical successes, multiparametric MRI and other GBCA-based MRI detection methods are the exception in the body owing to the lack of tumor specificity of the GBCA. There is thus an unmet need for targeted MRI contrast agents for accurate high-resolution detection and characterization of aggressive tumors to guide efficacious treatment at the earliest possible stage to improve the survival of cancer patients. An innovative design of a targeted MRI contrast agent specific to a cancer biomarker would have a significant impact to advance magnetic resonance molecular imaging (MRMI) for precision cancer imaging.
There have been continuous efforts to design and develop targeted MRI contrast agents since the clinical application of MRI in the early 1980s.5–9 Antibody targeted GBCAs were explored to target the biomarkers on cancer cell surfaces. Because of the low concentration of the cell-surface tumor markers (typically nM) and poor sensitivity of MRI for molecular imaging GBCA (typically μM), these targeted contrast agents have been unable to generate tumor-specific MRI detectable signal enhancement.5 Incorporation of tumor-specific antibodies and peptides into nanoparticles loaded with GBCAs deliver a sufficient amount of the contrast agents in solid tumors and produces robust signal enhancement for effective MRMI of cancer in animal tumor models.10 However, the large size of these agents impedes rapid target accumulation and clearance of GBCAs demanded of clinically safe and useful agents.11 Therefore, despite the efficacy of the targeted GBCA-nanoparticles in animal tumor models, they have not advanced to clinical development.
We have recently hypothesized that the design and development of small molecular targeted GBCAs specific to abundant oncoproteins in tumor extracellular matrix (ECM) would overcome the current limitations of MRI for cancer molecular imaging and precision cancer diagnosis.12 Tumors have a unique microenvironment enriched with oncogenic ECM proteins, not present in normal tissues. Among various ECM proteins in tumor tissues, extradomain B fibronectin (EDB-FN) is a promising target for designing clinically translatable targeted GBCA.13 Extradomain B fibronectin is an oncofetal isoform of FN and is highly expressed in the ECM of aggressive tumors.14 It is considered a marker of epithelial to mesenchymal transition which is a biological program associated with invasion, metastasis, and drug resistance of multiple malignancies.15–18 Elevated expression of EDB-FN is associated with epithelial to mesenchymal transition induction, cancer cell invasion, and metastasis.16 It is overexpressed in various aggressive human cancers and absent in normal tissues.19–21 Extradomain B fibronectin has been explored as an oncotarget for developing molecular imaging technologies and targeted therapeutics for cancer diagnosis and therapy. For example, antibody fragment L19 specific to EDB-FN has been used to develop positron emission tomography probes and targeted therapeutics for cancer imaging and therapy in both preclinical and clinical studies.22 A nanobody has been developed to target EDB-FN for effective positron emission tomography imaging of different types of cancer in animal tumor models.23 An aptamer-like peptide has also been developed to target EDB-FN to deliver imaging agents and therapeutics for cancer imaging and therapy.24
To encourage clinically and industrially acceptable properties, we used phage display against EDB fragment to develop a small peptide named ZD2 (Thr-Val-Arg-Thr-Ser-Ala-Asp) that has a moderate binding affinity to the EDB fragment.25 The peptide was conjugated to a clinical contrast agent gadoteridol (Gd(HP-DO3A) or ProHance) making ZD2-Gd(HP-DO3A) with a (CH2CH2O)2 spacer.25 The agent was effective for MRMI of aggressive tumor detection and characterization of aggressive prostate cancer and metastatic breast cancer in mouse models.26,27 The structure of the targeted contrast agent was further optimized to identify a lead targeted contrast agent ZD2-N3-Gd(HP-DO3A) (MT218, molecular weight of 1442.62 Da; Fig. 1) with an improved r1 relaxivity and a straightforward synthetic procedure for clinical translation.28 MT218 produces robust specific magnetic resonance (MR) contrast enhancement at doses lower than those of the base GBCA in several types of aggressive tumors, including breast cancer, colorectal cancer, head and neck cancer, pancreatic cancer, and prostate cancer.17,28–31 The enhancement pattern closely reflects relative EDB-FN expression levels in these tissue types, which is known to reflect tumor aggressiveness. MT218 in MRMI therefore has the potential for characterizing the invasiveness of solid tumor and assessing tumor response to therapies as well as development of drug resistance.17,32 MT218 is also a small, water soluble peptidic monomer with practical industrialization potential and rapid animal pharmacokinetics as required for commercial contrast agents.
 FIGURE 1:
FIGURE 1: Chemical structure of the targeted MRI contrast agent MT218.
In this work, we further investigated the dosing effect of MT218 for MRI of prostate cancer xenografts in rats and its potential for detection of lung cancer and pancreatic cancer models at a low dose in mice. We also performed a comprehensive in vitro characterization and in vivo pharmacokinetics, biodistribution, and safety assessment of MT218 according to the Food and Drug Administration (FDA) guidelines for preclinical development (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/developing-medical-imaging-drug-and-biological-products-part-1-conducting-safety-assessments).
MATERIALS AND METHODSThe MT218 active pharmaceutical ingredient (API) and MT218 injection were supplied by Jiangsu Ronghui Motek Pharmaceuticals, Inc (China). 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and diethylenetriaminepentaacetic acid (DTPA) were purchased from Strem Chemicals. 10-(2-Hydroxypropyl)-1,4,7,10-Tetraazacyclododecane-1,4,7-triacetic acid (HP-DO3A) was purchased from USP (Maryland). Human, rat, and dog plasma was obtained from Equitech-Bio, Inc (Kerrville, TX). Gadoteridol (ProHance, Ga(HP-DO3A)) was purchased from Bracco Diagnostics (Monroe Township, NJ). The pooled cryopreserved human primary hepatocytes, the CD1 mouse primary hepatocytes, the Sprague-Dawley (SD) rat primary hepatocytes, and the beagle dog primary hepatocytes were supplied by BIOIVT (Westbury, NY). The pooled human liver microsomes were purchased from BIOIVT. Recombinant HEK293 cell lines stably expressing human transporters (OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, and MATE2K), HEK293-MOCK cell line transfected with empty vector, human multidrug resistance protein 1 (MDR1) vesicles, and breast cancer resistance protein (BCRP) vesicles were supplied by GenoMembrane (Kanagawa, Japan). Human embryonic kidney cell line HEK-293 with stable expression of human ether-a-go-go related gene (hERG) was supplied by the Academy of Military Medical Sciences, China. Liver S9 microsomal fractions (S9) were provided by CHI Scientific (Mississippi). Histidine-deficient Salmonella typhimurium, TA97a, TA98, TA100, TA1535, and TA102, were obtained from Moltox (Boone, NC). Chinese hamster lung (CHL) fibroblast was obtained from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). Procedures carried out on the animals involved in the imaging experiments were performed according to an approved protocol by the Institutional Animal Care and Use Committee (IACUC) of Case Western Reserve University. All other animal studies were performed at Joinn Laboratories (Beijing, China) following the standard operating procedures and protocols approved by the IACUC and current Good Laboratory Practice (cGLP)/non-GLP guidelines of Joinn Laboratories.
Chelation StabilityAt pH 5 and 7.4, 0.25 mM MT218 was incubated with the same concentration of DOTA, DTPA, HP-DO3A, ethylenediaminetetraacetic acid (EDTA), or 0.50 mM zinc chloride and at room temperature or 37.5°C over a period of 30 days. Acetate buffer (0.05 M, pH 5) and Tris buffer (0.05 M, pH 7.4) were used to maintain the pH. A high-performance liquid chromatography–ultraviolet method (Agilent 2200, Waters Symmetry C18 column, 4.6 × 150 mm, 5 μm, mobile phase 0.08% trifluoroacetic acid acetonitrile/water) was used to analyze the remaining MT218 in the solution.
RelaxivitySolutions of MT218 were prepared at concentrations of 0.125, 0.25, 0.5, 1, and 2 mM in human serum albumin aqueous solution (40 mg/mL), water, and phosphate-buffered saline (PBS). The T1 and T2 values of the MT218 solutions were measured at 1.4 T using a Bruker Minispec Relaxometer (60 Hz, 37°C). The respective T1 and T2 values of MT218 solutions were also determined at 3 T using a 3 T MRS 3000 scanner (MR Solutions, Surrey, UK). T1 values were measured using an inversion recovery–fast low angle shot sequence from MR Solutions. T2 values were obtained using a multiecho multislice sequence from MR Solutions. The following equation is used to calculate relaxivities (r1, r2): 1/Ti = 1/Ti,0 + riC, where Ti is the observed relaxation time in the presence of the contrast agent, Ti,0 is the relaxation time of pure water, and C is the concentration of the MRI contrast agent. The subscripts indicate longitudinal (i = 1) or transverse (i = 2) relaxivities and relaxation times, respectively. The r1 and r2 relaxivities were calculated from the slopes of 1/T1 and 1/T2 versus MT218 concentration plots, respectively.
Binding Affinity StudyPlasmid DNA encoding EDB fragment was constructed with a T7 promoter and a decameric His-tag at the N-terminal. A Lac operon (lacO) was also incorporated to control EDB expression with Isopropyl β-d-1-thiogalactopyranoside (IPTG) induction. The recombinant EDB expressing plasmid was transformed into Escherichia coli strain BL21(DE3)-T1, and EDB expression was induced by the addition of 1 mM IPTG at the midlog phase of bacterial growth and incubated for 3 hours. The bacteria were collected and lysed with lysozyme. Extradomain B was purified using a HisTalon SuperFlow cartridge and lyophilization. The size and purity of the extracted EDB protein were determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Extradomain B protein protein fragment (5 mg/mL) was further purified with a PD-10 desalting column using pH 7.4 10 mM PBS buffer as the mobile phase.
The binding affinity of MT218 to EDB protein fragment was determined using surface plasmon resonance. Stock solution of MT218 (1.0 mM) was prepared with a running buffer (0.05% Tween-20 in 10 mM PBS, pH 7.4). The active and reference flow cells of a CM5 sensor chip (Cytiva, Sweden) were activated with amine coupling kit, and EDB protein (25 μg/mL) in 10 mM acetate buffer (pH 4.5) was injected into the active cell for 3 minutes at a rate of 10 μL/min to immobilize the protein at a level of 1500 RU. Then both the reference and flow cells were blocked with ethanolamine (pH 8.5) for 7 minutes. The system was then switched to running buffer, and varying concentrations of MT218 were injected for 120 seconds at 50 μL/min, with 5 minutes of dissociation. Binding affinity was determined using Biocore T200 Evaluation Software 3.1. Microsoft Office Excel (2019) was used for statistical analysis of data, including mean, standard deviation, and coefficient of variation (CV %).
Plasma Protein BindingNonspecific binding of MT218 to plasma proteins was determined by incubating with human serum albumin (40 mg/mL), human, dog, rat plasma, and PBS at 37°C for 3 minutes at concentrations of 50, 80, and 110 μM, respectively. After incubation, MT218 (or gadolinium [Gd]-containing degradants) was separated by centrifugation using a 10 kDa cut-off filter at the speed of 14,000g at 4°C for 30 minutes. Filtrate (300 μL) from each sample tube was transfered to a 15-ml metal free Eppendorf tube, and 9.7 ml of 3% nitric acid was then added. The mixture was kept overnight. Then the samples were centrifuged to remove insolubles and tested by inductively coupled plasma-optical emission spectrometry (ICP-OES 730, Agilent, Santa Clara, CA after agilent) for Gd content. The total MT218 binding to plasma proteins was calculated into the binding ratio.
Plasma StabilityMT218 (0.5 mM) was incubated in human, rat, and dog plasma at 37°C for 48 hours. Phosphate-buffered saline was used as control. The mixture was mixed with methanol, followed by centrifugation, and then measured using analytic high-performance liquid chromatography (Agilent 1260 Infinity DAD LC, column Phenomenex C18, 4.6 × 250 mm, 5 μm; phase A: 0.08% TFA·H2O; phase B: 0.08% trifluoroacetic acid in acetonitrile; 0–13 minutes phase B, 0%–13%). The percentage ratio of remaining MT218 was calculated.
MetabolismIn a series of non-GLP studies, metabolic stability, metabolite identification, and possible metabolic pathway of MT218 were investigated by incubation with hepatocytes from human, CD1 mouse, SD rat, beagle dog, and cynomolgus monkey following FDA guidance on in vitro drug interaction studies. Testosterone and midazolam (substrates of CYP3A4) and 7-hydroxycoumarin (substrate of uridine 5′-diphospho-glucuronosyltransferase) were used as positive controls. The inhibition effect of MT218 on main members of cytochrome P450 (CYP450) family, including CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, was evaluated using pooled human liver microsomes at the MT218 concentration from 0.01 to 100 μM. The final concentration of the human liver microsomes was 0.5 mg/mL. The induction potential on CYP1A2, CYP2B6, and CYP3A4 was also investigated using omeprazole (50 μM), phenobarbital (1000 μM), and rifampicin (25 μM) as positive control. The potential inhibition of MT218 on solute carrier family transporters MDR1, BCRP, organic anion transporting polypeptides (OATP1B1, OATP1B3), organic anion transporters (OAT1, OAT3), organic cation transporter 2 (OCT2), and multidrug and toxin extrusion proteins (MATE1 and MATE2K) was evaluated. The inhibition potential of MT218 on human MDR1 and BCRP was assessed using membrane vesicles expressing MDR1 and BCRP, respectively, according to the FDA's guidance (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions). For all the other transporters, HEK293 cells expressing these transporter proteins were used. Cyclosporine A (20 μM), novobiocin (100 μM), rifampin (100 μM), quinidine (500 μM), probenecid (50 μM), and pyrimethamine (10 μM) were used as inhibitors, respectively. The MT218 concentrations were 100, 50, 10, 5, and 1 μM. The samples were analyzed using the Shimadzu LC20 liquid chromatography (Shimadzu Co, Ltd, Japan) system and API 4000 triple-quadrupole mass spectrometer (Applied Biosystems Inc).
Cell CultureLNCaP and PC3 human prostate cancer cell lines and H1299 human lung cancer cell line were purchased from ATCC (Manassas, VA). The cell lines were cultured in RPMI 1640 medium (Gibco, Waltham, MA) supplemented with 10% fetal bovine serum and 100 units/mL penicillin/streptomycin. All the cells were grown at 37°C and 5% CO2. KPC-K8484 (herein KPC) cells were generously provided by the lab of Dr Jordan Winter (University Hospitals of Cleveland and CWRU, Cleveland, OH). KPC cells were maintained in RPMI 1640 Medium supplemented with 10% fetal bovine serum (Sigma-Aldrich, St Louis, MO) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA).
Western BlotTotal protein was extracted from cancer cells as previously described.16 Protein concentration was determined by Lowry assay. Equal amounts of protein extracts (40 μg) were loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis for electrophoresis and transferred onto nitrocellulose membranes. The following primary antibodies (1:1000 dilution, overnight incubation at 4°C) were used: anti-β-Actin (Cell Signaling Technology, Danvers, MA) and anti-EDB-FN (G4 clone, Absolute Antibody, UK). Following secondary antibody incubation (1:2000 dilution for 2 hours), the membranes were developed using Signal Fire Plus ECL Kit (Cell Signaling Technology) and imaged on ChemiDoc XRS+ Imager (Bio-Rad, Hercules, CA).
Rat Tumor Xenograft Mouse ModelsAdult male NIH-Foxn1rnu nude rats were obtained from Charles River Laboratories (Wilmington, MA). The rats were housed in the Animal Facility at CWRU and all animal experiments were performed according to the protocols approved by IACUC. The lateral flanks of 10 rats were subcutaneously injected with either 4 × 106 PC3 or 8 × 106 LNCaP cells suspended in a Matrigel-PBS mixture (1:1). Once the tumor volume exceeded 100 mm3, rats were subjected to MRI. A total of 20 male rats were used to develop subcutaneous xenografts of human prostate cancer. Ten rats were subcutaneously implanted with either PC3 or LNCaP human prostate cancer cells. Six rats developed visible PC3 tumors within 5 weeks and only 3 rats developed LNCaP tumors within 6 weeks.
Mouse Tumor Xenograft Mouse ModelsFor the protein kinase C (PKC) pancreatic cancer model, C57BL/J6 immunocompetent mice (6-week-old males and females) were purchased from the Jackson Laboratory (Bar Harbor, MA). KPC-K8484 cells (1 × 105) suspended in Matrigel-PBS mixture (1:1) were subcutaneously injected into the left flank of the B6 mice (100 μL per mouse, 5 mice per group). Magnetic resonance molecular imaging with MT218 was performed at 1 and 5 weeks after tumor inoculation. For lung cancer model, nude athymic mice (6-week-old nu/nu males) were purchased from Jackson Laboratory. About 5–6 × 106 H1299 cells suspended in Matrigel-PBS mixture (1:1) were subcutaneously injected in the left flanks of the mice. The mice were housed and cared in the Animal Facility at CWRU.
MRI With MT218Magnetic resonance imaging was performed on a 3 T MRS 3000 scanner (MR Solutions) with a mouse short quad coil or a rat short quad coil (MR Solutions) with respiratory gating. The animals were anesthetized with isofluorane and tail vein catheter was setup. For mouse MRI, T1-weighted MR images were obtained before and after injection of MT218 at a pre-determined dose with axial fast spin echo sequence (repetition time, 305 milliseconds; echo time, 11 milliseconds; flip angle, 90 degrees; field of view, 40 mm × 40 mm; slice thickness, 1 mm; slice number = 15; number of average, 2; matrix, 256 × 256). For rat MRI, T1-weighted images were acquired before and after injection of MT218 at doses of 0.04 or 0.02 mmol/kg, using a fast spin-echo sequence with respiratory gating (repetition time, 305 milliseconds; echo time, 11 milliseconds; field of view, 80 × 80 mm; slice thickness, 1 mm; number of average, 2; matrix, 128 × 128). The imaging protocol was repeated with the clinical agent ProHance as a control at the clinical dose of 0.1 mmol Gd/kg. Because the contrast-enhanced MRI experiments were noninvasive, the same rats with tumor xenografts were used for testing different agents and doses. All the imaging studies applied the crossover design, with a washout period of at least 3 days. The experiments with MT218 at the dose of 0.04 mmol/kg were performed first, followed by 0.02 mmol/kg MT218 or gadoteridol at 0.1 mmol/kg for the second or third MRI. All animals were euthanized immediately after the last scan.
Magnetic resonance data were exported into digital imaging and communication in medicine files and analyzed using FIJI software. The slice of the tumor was selected based on the contrast levels of the enhanced image. Circular regions of interest (ROIs) of 10 to 20 mm2 were drawn in the area of the muscle. Another ROI was drawn outlining the tumor. Mean signal intensity in the ROIs was measured in the images. The contrast-to-noise ratio (CNR) in the tumors was calculated using the following equation, CNR=Mean IntensityTumor−Mean IntensityMuscleσnoise,where σnoise is the standard deviation of intensities from an ROI in the lower left corner of the image. The CNR of tumors was calculated before and at 10, 20, and 30 minutes postinjection.
Immunohistochemical StainingAfter the last MR image acquisition, the animals were euthanized and tumor tissues were collected for histological and immunohistochemical analyses. Tissue fixation, sectioning, hematoxylin and eosin, and immunohistochemistry staining were performed at the tissue resource core of Case Western Reserve University (Cleveland, OH). Anti-EDB-FN antibody, G4 (Absolute Antibody, Boston, MA), diluted in PBS solution with Tween-20 (PBS-T, 1:100) was applied to the tissue and incubated for 1 hour. Primary antibody detection was performed with Rabbit on Rodent HRP Polymer detection solution for 30 minutes (Biocare Medical, Pacheco, CA). Images were acquired using an Olympus Bx61VS slide scanner (Olympus America, Center Valley, PA) and processed in OlyVIA software.
The Safety PharmacologyMT218 was evaluated by Eurofins Scientific (San Diego, CA) across a panel of in vitro SAFETYscan E/IC50 assays for potential biological activities in a screen of 78 common biological targets (ADORA2A, ADRA1A, ADRA2A, ADRB1, ADRB2, AVPR1A, CCKAR and CNR1 agonist and antagonist, etc.) over the concentration range of 0.005 to 100 μM following Eurofins' internal standard operating procedures.33 The assays were performed utilizing the PathHunter enzyme fragment complementation technology, FLIPR-based cellular screening assays, KINOMEscan kinase binding assays, and a variety of enzymatic assays. Positive reference compounds, including NECA and SCH 442416 for ADORA2A calcium flux agonist and antagonist, 61603 hydrobrimide and tamsulosin for ADRA1A calcium flux agonist and antagonist, and UK 14304 and yohimbine for cAMP agonist and antagonist, were assayed in parallel to verify acceptable performance of the assays.
GLP-compliant studies were performed to evaluate the effects of MT218 on the hERG currents, CNS functions in SD rats, and cardiovascular and respiratory system functions in conscious beagle dogs. For the hERG experiment, a Multi Clamp 700B manual patch clamp system (Axon Instruments) was used to induce hERG currents of human embryonic kidney cells HEK293 cell in whole-cell voltage clamp mode. MT218 at 1, 3, 10, 30, and 100 μM or positive control terfenadine at 10 μM was tested. The hERG channel tail currents and the peaks of the tail currents were recorded by Clampex 10.3, and the data were analyzed by Clampfit 10.3.
To evaluate the effects on CNS, 4 groups of (5/sex/group) female and male SD rats received single intravenous (IV) injection of 0, 0.1, 0.3, and 1 mmol/kg. Animals were tested by the functional observational battery assay at 0.25, 0.75, 1.5 and 4 hours after dosing, including home cage observations, hand-held observations, open-field observations, responses to a variety of stimulus, and measurements for grip strength (forelimb) and body temperature. The effects of MT218 injection on cardiovascular and respiratory system functions were explored in conscious beagle dogs following a single IV injection of saline control, MT218 at 0.07, 0.21, and 0.70 mmol/kg in a total of 9 dogs (4 males and 5 females). The dose volume of each animal was 3.5 mL/kg. Electrocardiographic parameters (heart rate, PR interval, QRS duration, QT and QTcF interval, QRS voltage, STe, heart rhythm, and electrocardiogram [ECG] waveform analysis), blood pressure (systolic blood pressure, diastolic blood pressure and mean arterial blood pressure), respiratory frequency, and tidal volume were recorded, analyzed, and evaluated within 1 hour before dosing and at 0.5, 1, 2, 4, and 24 hours after dosing.
PharmacokineticsThe pharmacokinetics of MT218 was studies in SD rats and beagle dogs after single IV injection of 0.04, 0.1, and 0.2 mmol/kg of MT218 (3 animals/sex). For rats, blood samples were collected from jugular veins at predose and at 2, 5, 10, 15, 30, and 45 minutes and 1, 2, 3, and 4 hours after dosing from group 1 to 3. For dogs, blood samples were collected from fore limb veins at predose and at 5, 10, 15, 30, and 45 minutes and 1, 1.5, 2, 3, 4, and 6 hours after dosing from group 1 to 3. A validated inductively coupled plasma mass spectrometric method (ICP-MS, Agilent 7800) was used for quantitation of Gd concentration in the plasma. Terbium (Tb) was used as internal standard. Plasma samples (50 μL) and 10 μL IS solution (Tb, 5 μg/mL) were mixed. Then ultrapure water-65% nitric acid-triton X-100 (100:0.1:0.1, v/v/v) was added to dilute samples before analyzed by ICP-MS. Pharmacokinetic parameters, such as half-life T1/2, concentration immediately after dosing (C0), area under the curve (AUC), volume of distribution (Vz), clearance (Cl), mean residence time, and AUC ratio, were calculated by a noncompartment model (noncompartmental analysis) using WinNonlin software (version 8.0).
BiodistributionFor the biodistribution, 30 rats (3/sex/group) received a single IV injection of MT218 at 0.1 mmol/kg. Blood samples and tissues were collected before dosing and 10 and 30 minutes and 2, 6, and 48 hours after dosing. The tissues collected were heart, liver, spleen, lung, kidney, stomach, small intestine, gonads, brain, fat, muscle, submaxillary lymph nodes, prostate (males), skeleton and abdominal skin. Six animals from each group (groups 1–5) were used at each time point. The tissues were homogenized and then digested with nitric acid in Water Bath Constant Temperature Oscillator (Taicang Experimental Equipment Factory & Suzhou Peiying Experimental Equipment Co Ltd). A validated ICP-MS method (Agilent 7800) was used for quantitation of Gd in the rat plasma and tissues. The metabolic parameters were calculated by the noncompartmental analysis using WinNonlin software (version 8.0).
ExcretionThe urine, feces, and bile excretion of MT218 was investigated in SD rats after IV injection of 0.1 mmol/kg in a non-GLP study. One group (3/sex) of rats was used for assessing the excretion of the Gd chelate into urine and feces. The feces and urine samples were collected before dosing (D-1) and at 0–4, 4–8, 8–24, 24–48 (D1–D3), 48–72 (D3–D4), 72–96 (D4–D5), 96–120 (D5–D6) and 120–144 (D6–D7) hours after dosing. The second group was used for assessing bile excretion using conscious biliary cannulation. The bile was collected 30 minutes predose and 0–1, 1–2, 2–4, 4–6, 4–6, and 6–8 hours after dosing. Gd content in the samples was determined by ICP-MS (Agilent 7800).34
ToxicitySingle and repeated toxicity was performed under GLP conditions in SD rats and beagle dogs. Rats were 6 to 7 weeks old and weighed 200 to 246 g for males and 175 to 224 g for females on day -1 (Beijing Vital River Laboratory Animal Technology Co, Ltd). Male dogs were 6 to 7 months old and weighed 7.34 to 10.58 kg. Female dogs were 6 to 7 months old and weighed 6.50 to 9.87 kg on day −1 (Marshall Biotechnology [Gu'an] Co, Ltd). The toxicity, toxicokinetic profile, and potential target organs' toxicity and tissue distributions of MT218 were investigated after a single IV injection at a dose of 1.39 mmol/kg or daily IV injection for 2 weeks at doses of 0.15, 0.46, and 1.39 mmol/kg/d in rats; and at a single dose of 0.70 mmol/kg or daily injection at 0.07, 0.21, and 0.70 mmol/kg/d for 2 weeks in dogs according to the study design listed in Table 1. Saline injection was used as a placebo control. The animals were observed during and after the injections for potential side effects. The reversibility of any potential toxicity was monitored during an additional 2-week recovery period. Parameters evaluated in the study included dose formulation analysis, mortality and moribundity, clinical observations, body weights, food consumption, body temperature, ophthalmoscopic examinations, hematology, coagulation, clinical chemistry, urinalysis, plasma drug concentration, and toxicokinetic analysis. At the end of the recovery period, the animals were euthanized, and tissues and organs were collected and weighed and then subjected to macroscopic and microscopic histological examinations for any sign of toxic side effects. Rat tissues of the bone (femur), skin (abdomen), liver, brain, muscle (quadriceps femoris), lung, heart, kidney, spleen, large intestine (colon), and small intestine (jejunum) were collected at scheduled necropsy on day 29 for groups 1 to 4 and day 15 for single-dose group 5 to test the Gd remaining in the tissues using a well-established ICP-MS method. The no observed adverse effect level (NOAEL) was determined as the highest dose at which there are no biologically significant increases in the frequency or severity of adverse effect between the test and control groups.
TABLE 1 - Design and Group Assignment of Rat and Dog Toxicity Studies Group Control/MT218 Dose Level, mmol/kg/d Number of Animals/Sex Notes Rat toxicity 1 Control 0 5 + 10 + 5* Repeated dose 2 MT218 0.15 10 + 5† 3 MT218 0.46 10 + 5† 4 MT218 1.39 10 + 5† 5 MT218 1.39 5 + 5‡ Single dose 6 Control 0 4 + 2§ Toxicokinetics 7 MT218 0.15 8 + 2§ 8 MT218 0.46 8 + 2§ 9 MT218 1.39 8 + 2§ Dog toxicity I Control 0 3 + 3 + 2∥ Repeated dose II MT218 0.07 3 + 2¶ III MT218 0.21 3 + 2¶ IV MT218 0.70 3 + 2¶ V MT218 0.70 3 + 2# Single dose*The first 5 animals per sex were designated for the necropsy on day 3, the middle 10 animals per sex were designated for the necropsy after 2 weeks of dosing, and the last 5 animals per sex were designated for necropsy after the 2 week recovery period following the last dosing.
†The first 10 animals per sex per group in groups 2–4 were designated for the necropsy after 2 weeks of dosing. The last 5 animals per sex per group were designated for necropsy after the 2-week recovery period after the last dosing.
‡The first 5 animals per sex were designated for the necropsy on day 3 and the last 5 animals per sex per group were designated for necropsy on day 15.
§The last 2 animals per sex per group were included as TK replacement animals and received dosing. They were euthanized after the last blood sample collection.
∥The first 3 animals per sex were designated for the necropsy on day 3, the next 3 animals per sex were designated for the necropsy after 2 weeks of dosing on day 15, and the last 2 animals per sex were designated for necropsy after the 2-week recovery period after the last dosing on day 29.
¶The first 3 animals per sex per group were designated for necropsy after 2 weeks of dosing. The rest 2 animals per sex per group were designated for necropsy after the 2-week recovery period after the last dosing.
#The first 3 animals per sex were designated for the necropsy on day 3 and the remaining 2 animals per sex per group were designated for necropsy on day 15.
Genotoxicity of MT218 was evaluated using a S. typhimurium mutagenicity test, a chromosomal aberration test in CHL fibroblasts, and in vivo, a micronucleus assay in Institute of Cancer Research (ICR) mice. The mutagenicity of MT218 injection was evaluated in TA97a, TA98, TA100, TA102, and TA1535 at the dose level of 15, 50, 150, 500, 1500, and 5000 μg/plate. The vehicle control (sodium chloride injection); positive controls of direct mutagens 9-aminoacridine (50 μg/plate), 2-nitrofluorene (10 μg/plate), sodium azide (6 μg/plate), and 4-nitroquinoline N-oxide (1 μg/plate); and indirect mutagens 2-aminoanthracene (3 μg/plate) and benzopyrene (5 μg/plate) were also included in the test. The potential genotoxicity of MT218 injection to induce chromosomal aberration in CHL fibroblast with or without rat liver S9 metabolic activation was evaluated at 2000, 1000, and 500 μg/mL for 4 or 27 hours. The vehicle control (sodium chloride injection) and positive controls of direct clastogen mitomycin C at 0.1 μg/mL and indirect clastogen cyclophosphamide monohydrate at 10 μg/mL were also included in the test. One hundred or 300 well-spread metaphase cells per treatment were evaluated, and chromosomal aberration ratio for each treatment was calculated.35 The micronucleus assays in ICR mice were tested in a total of 32 male ICR mice (Vital River Laboratories Co, Beijing, China), with 8 mice per group in the 2000 mg/kg/d group and 6 mice per group in the vehicle control and 500 and 1000 mg/kg/d test groups. Cyclophosphamide monohydrate was used as positive control. The animals of all groups were dosed once daily for 2 days, and bone marrow was collected analyzed at about 22.5 hours after the final dose from all animals.35
Other GLP-compliant toxicology studies included an in vitro hemolysis test for MT218 using rabbit red blood cells (RBCs) and assessment of the potential for active systemic anaphylaxis of MT218 using guinea pigs. For the in vitro hemolysis test, 0.1, 0.2, 0.3, 0.4, and 0.5 mL of MT218 at 0.2 mol/L was incubated with 2.5 mL of 2% rabbit RBC suspension at 37°C ± 0.5°C, with saline and sterile water as negative and positive control. Hemolysis and aggregation were recorded at 15, 30, and 45 minutes and 1, 2, and 3 hours after incubation. For the potential active systemic anaphylaxis, a total of 36 male guinea pigs were randomly assigned to 4 groups (9 animals/group): saline group, human albumin group (40 mg/kg for sensitization, and 80 mg/kg for challenge), and 2 MT218 dose groups (0.200 and 0.695 mmol/kg for sensitization and doubled concentrations for challenge.) Animals were sensitized by intraperitoneal injection on days 1, 3, and 5. Fourteen days after last sensitization injection (day 19), the first 3 animals of each group received foot IV injection challenge. The rest animals of each group were challenged 21 days after last sensitization injection (day 26). Animals were observed twice daily (am and pm) during the study period for signs of mortality, mental state, behavior, morbidity, respiration, secretion, feces, and capability of water and food intake. Body weights were recorded before, during, and at the end of the study.
RESULTS Relaxivity and Binding Affinity of MT218The r1 and r2 relaxivities of MT218 are listed in comparison with r1 relaxivity of gadoteridol in Table 2. The r1 relaxivity of MT218 was nearly 2-fold of that of gadoteridol, but the r1/r2 ratio is similar to gadoteridol. The relaxivities did not change substantially when the magnetic field strength increased from 1.4 to 3.0 T. The results were the same as for the previously reported relaxivities.26 The r1 relaxivity of MT218 was also significantly higher than that of the prototype agent ZD2-Gd(HP-DO3A) (4.12 mM−1 s−1 at 1.4 T) that used a (CH2CH2O)2 spacer. The increased relaxivity might be attributed to the more rigid spacer, a 5-membered triazole ring, between the peptide and Gd(HP-DO3A), which probably deceases the rotational rate of the chelate.38,39 The binding affinity constant (Kd) of MT218 to the EDB fragment was measured in the range from 0.76 to 6.88 μM, with the average binding affinity of 3.45 μM (n = 4). The moderate binding affinity enables a rapid binding of the agent to the ECM oncoprotein at the clinically expected GBCA and EDB-FN concentrations and would allow reversible binding of the agent to facilitate rapid clearance from the body after imaging.
TABLE 2 - Relaxivities of MT218 Contrast Agent Magnetic Field r 1 Relaxivity, mM−1 s−1 r 2 Relaxivity, mM−1 s−1 Gadoteridol 1.4 T36 3.2 (H2O) 1.5 T37 2.9 (H2O) 3.2 (H2O) 4.1 (plasma) 5.0 (plasma) 3.0 T37 2.8 (H2O) 3.4 (H2O) 3.7 (plasma) 5.7 (plasma) MT218 1.4 T 5.34 (H2O) 7.40 (H2O) 6.58 (PBS) 8.87 (PBS) 6.54 (HSA) 8.70 (HSA) 3.0 T 6.25 (H2O) 7.90 (H2O) 6.16 (PBS) 7.91 (PBS)PBS indicates phosphate-buffered saline; HSA, human serum albumin.
The Gd(III) chelation stability of MT218 was determined by challenging the chelate with different chelating ligands and Zn2+ ions.40 MT218 is highly stable, at least kinetically, at neutral pH 7.4 and no transmetallation or dissociation of MT218 was observed in the presence of DOTA, DTPA, HP-DO3A, EDTA, and Zn(II) at pH 7.4 and the temperature of 25°C and 37.5°C for 1 month (Fig. 2A). At pH 5 and 25°C, MT218 also showed chelation stability for at least 30 days. MT218 was stable at pH 5 and 37.5°C in acetate buffer (Fig. 2B). Loss of MT218 was observed at pH 5 and 37.5°C in the presence of the free chelating ligands, DOTA, DTPA, HP-DO3A, and EDTA, probably because of competitive acid-assisted dissociation of the Gd(III) from MT218 (Fig. 2C). Nevertheless, the presence of Zn(II) ions did not cause dissociation of MT218 at pH 5 and 37.5°C for at least 30 days (Fig. 2D).
 FIGURE 2:
FIGURE 2: Percentage of MT218 remaining in the mixture solution under the challenge of ZnCl2, DOTA, DTPA, HP-DO3A, and EDTA at pH 5 or 7.4 and a temperature of 25°C or 37.5°C, respectively. A, pH 5, 25°C, (B) pH 7.4, 25°C, (C) pH 5, 37.5°C, (D) pH 7, 37.5°C. The final concentration of MT218, DOTA, DTPA, HP-DO3A, and EDTA in the mixture solution was 0.25 mM. The final concentration of ZnCl2 in the mixture solution was 0.50 mM.
Plasma Protein Binding and StabilityAfter a short incubation (3 minutes) of MT218 with rat, dog, and human plasma, the binding ratios of the agent to plasma proteins was determined by the concentration of the bound MT218 to the total concentration in the solutions. The binding ratios were 28.44%, 27.12%, and 25.68% in rat plasma, 29.10%, 26.91%, and 48.44% in dog plasma, and 47.31%, 45.15%, and 40.50% in human plasma at concentrations of 50, 80, and 110 μM, respectively (n = 4). To test the stability of the peptide in MT218, the agent was incubated in PBS and rat, dog, and human plasma at 37°C for up to 48 hours. After incubating MT218 for 1, 3, 6, 28, and 48 hours, the average remaining percentage of intact MT218 was 74.87%, 20.54%, 4.61%, 0%, and 0% in rat plasma; 89.19%, 71.45%, 47.81%, 11.23%, and 5.38% in dog plasma; and 83.47%, 48.79%, 12.87%, 5.21%, and 3.97% in human plasma, respectively (Fig. 3) (n = 2). The enzymatic degradation half-life (t1/2) of MT218 was 1.63, 5.85, and 2.63 hours in rat, dog, and human plasma, respectively. The results indicate that the ZD2 peptide in MT218 undergoes enzymatic cleavage by peptidases in plasma. The peptide degr
留言 (0)