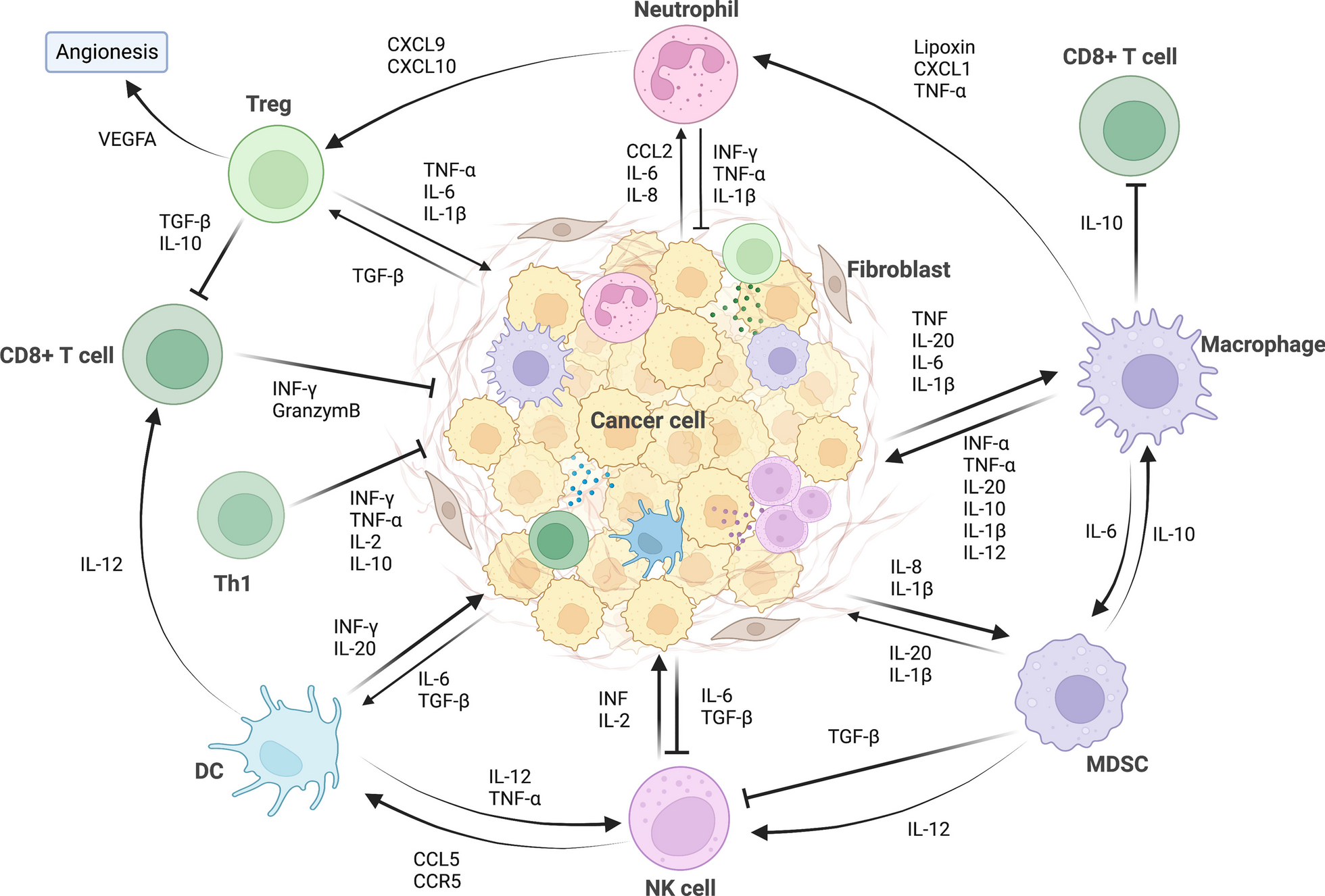
In this section, we discuss ongoing strategies targeting the myeloid compartment in the preclinical and clinical settings which include: (1) altering myeloid cell composition within the TME through enhanced differentiation, proliferation, and recruitment of myeloid cells; (2) functional blockade of immune-suppressive myeloid cells; (3) reprogramming via either polarization, metabolic, or epigenetic modification of myeloid cells to acquire pro-inflammatory properties; (4) modulating myeloid cells via cytokines; (5) myeloid cell therapies; and (6) emerging targets such as Siglec-15, triggering receptor expressed on myeloid cells 2 (TREM2), macrophage receptor with collagenous structure (MARCO), leukocyte immunoglobulin-like receptor B2 (LILRB2), and common lymphatic endothelial and vascular endothelial receptor 1 (CLEVER-1) (Table 1).
Strategies to alter myeloid cell differentiation, proliferation, and recruitment with the tumor microenvironment
In response to tumor-derived factors, immunosuppressive myeloid cells are consistently recruited, expanded, or differentiated to fuel tumor progression. One of the most straightforward strategies of targeting myeloid cells for cancer treatment is to alter the myeloid population composition, reducing the pro-tumor myeloid cell infiltration and increasing the abundance of anti-tumor immune cells. Strategies ranging from chemoattractant blockade to myeloid growth factors have been studied extensively in both preclinical animal models and clinical trials.
CCL2–CCR2 axis
The CCL2–CCR2 plays an integral role in the recruitment of myeloid cells including inflammatory monocytes, TAMs, and MDSCs. In metastatic CRC models, liver metastases which contain TAMs with high CCR2 expression are linked to a worse prognosis [165]. Inhibition of the CCL2–CCR2 axis suppresses tumor metastasis through reduced angiogenesis in preclinical models, in both direct manner, since CCL2 itself exerts an angiogenic effect, and indirect manner, which is through reduced chemoattraction of monocytes and macrophages [166,167,168]. A variety of inhibitors have been studied in the clinical setting to assess tumor response, which are summarized below.
Carlumab (CNTO 888) is a human monoclonal anti-CCL2 antibody with primarily negative clinical results. Carlumab was ineffective as monotherapy, as seen in a phase II study (NCT00992186) involving second-line therapy for metastatic castrate-resistant prostate cancer, where the objective response rate (ORR) was 0% and the median progression-free survival (mPFS) was only 2.7 months [135]. However, carlumab in combination with conventional chemotherapy (docetaxel, paclitaxel, carboplatin, gemcitabine, and PEGylated liposomal doxorubicin) for advanced solid tumors demonstrated improved clinical responses, including an ORR of 37.5% and a duration of response (DOR) of 6.3 months [169, 170]. Unfortunately, the effects of carlumab may be short-lived based on median CCL2 serum concentrations collected throughout the study period. While there was an initial reduction in total levels at the two-hour mark following initiation, there was a subsequent threefold to fivefold increase with further doses compared to baseline, regardless of the chemotherapy backbone, suggesting chemotherapy alone may have resulted in tumor response. Based on safety data, carlumab is well tolerated with the chemotherapy, with the most common grade 3 treatment-related adverse events (TRAEs) being cytopenias, fatigue, and stomatitis.
PF-04136309 is a small-molecule oral CCR2 inhibitor. In two small phase I trials (NCT01413022 NCT02732938), PF-04136309 was added to chemotherapy (FOLFIRINOX, nab-paclitaxel, and gemcitabine) in the management of advanced pancreatic cancer and produced response rates ranging from 23.8 to 48.5% [171, 172]. Pulmonary toxicity was reported in 24% when PF-04136309 was combined with nab-paclitaxel and gemcitabine. In the exploratory analysis, almost all recipients of PF-04136309 were found to have a decrease in peripheral blood CD14+ CCR2+ monocytes, though CCR2+ TAMs remained present in the majority of biopsy samples.
BMS-813160 is a small-molecule inhibitor that antagonizes both CCR2 and CCR5 and is currently under investigation in combination with nivolumab for the treatment of a variety of tumor types (NCT03496662, NCT03767582, NCT03184870, NCT04123379, and NCT02996110). Neither carlumab nor PF-04136309 has ongoing trials at this time.
CSF-1R
CSF-1 is a major lineage regulator and chemoattractant for TAMs. Preclinical data have demonstrated that inhibition of CSF-1R signaling repolarizes TAMs from M2-like to M1-like anti-tumor phenotype rather than simply depleting TAMs [173, 174]. One issue encountered with CSF-1R blockade has been compensatory upregulation of PD-L1 and CTLA-4 to maintain tolerogenic abilities, so clinical models have focused on a dual inhibitory approach involving CSF-1R blockade and ICIs to overcome this effect [175, 176].
While antagonists like sunitinib grossly block class III receptor tyrosine kinases (c-KIT, FLT3, CSF-1R, and PDGFR), dedicated CSF1R inhibitors have been developed, including small-molecule agents (pexidartinib, ARRY-382, BLZ945, and vimseltinib) and monoclonal antibodies (emactuzumab, cabiralizumab, IMC-CS4, AMG820, lacnotuzumab, PD-0360324, and axatilimab) [177, 178]. However, few have been able to demonstrate meaningful clinical activity. Two phase I trials involving LY3022855 monotherapy (NCT02265536, NCT01346358) and one phase I trial involving AMG 820 (NCT01444404) in the management of advanced solid tumors reported zero objective responses (0/86 and 0/25, respectively), though decreases in TAMs were noted in addition to elevations in circulation CSF-1 levels, indicating that proper target engagement occurred [179,180,181]. When LY302285 is used in combination with ICIs including tremelimumab (anti-CTLA-4) or durvalumab (anti-PD-L1), ORR approaches 4.2% (3/72) [182]. Similarly, for AMG820, when combined with pembrolizumab (anti-PD-1) for advanced solid tumors, ORR has been documented at 2.6% (3/116), well below expected response rates seen with pembrolizumab monotherapy [183].
One area of promise for CSF-1 inhibitors is in the management of tenosynovial giant cell tumors (TGCTs) and pigmented villonodular synovitis (PVNS) which are both rare, nonmalignant tumors that originate from the synovium of musculoskeletal joints and occur because of CSF-1 overexpression due to CSF-1/COL6A3 translocations [184]. Pexidartinib received FDA approval in 2019 following the results of the phase III trial (ENLIVEN) which randomized patients with unresectable TGCTs to receive pexidartinib vs. placebo. Following a 25-week follow-up period, the ORR was 38% (vs. 0% placebo, p < 0.0001) with a complete response (CR) rate of 15% [185]. Interestingly, ORR rates were similar between placebo crossovers and the initial pexidartinib arm, with crossover participants experiencing less hepatotoxicity, so the FDA did not include a loading dose in the approval [186]. Unique adverse events reported in ENLIVEN included changes to hair color (67%), transaminitis (39%), and nausea (38%), and both periorbital (13%) and peripheral (13%) edema among others.
CXCR1/2
The release of IL-8 by malignant cells and its subsequent binding to CXCR1 and CXCR2 on circulating myeloid cells and surrounding endothelial cells leads to the recruitment of MDSCs to the TME and the promotion of angiogenesis [187]. Ibuprofen inhibits IL-8 signaling, both through cyclooxygenase-2 (COX2) and non-COX2 pathways, and has been used as a base model for the development of novel CXCR1/2 inhibitors, including reparixin and ladarixin [188]. Other backbones have been explored as well, including nicotinamide antagonists (SX-682) and thiazolopyrimidine derivatives (AZD 5069).
Reparixin showed promising single-arm phase I trial data when combined with weekly paclitaxel in metastatic HER2-negative breast cancer (ORR 30%) [189]. However, subsequent randomized, two-arm data from the phase II fRIDA trial failed to detect a difference in the primary endpoint of mPFS when comparing the combination therapy to paclitaxel alone (5.5 vs. 5.6 months, respectively) [190]. Ladarixin is a second-generation dual inhibitor with stronger affinity for CXCR2, slowed melanoma progression in preclinical models, but clinical trials remain absent at this time [191]. Ongoing trials involving allosteric, reversible, small-molecule inhibitors SX-682 and navarixin as monotherapies and in combination with PD-1/PD-L1 agents are currently underway (NCT04245397, NCT03161431, NCT04599140, NCT04477343, NCT04574583, and NCT03473925).
Indirect methods of CXCR1/2 inhibition are also emerging, including the development of monoclonal antibodies which bind and sequester IL-8, such as HuMax-IL8 (BMS-986253) which has been shown in preclinical models to reduce PMN-MDSCs and prevent the mesenchymalization of TNBC [192]. Following a phase I study, HuMax-IL8 was found to provide no objective response as monotherapy, but multiple follow-up trials are ongoing involving its use in combination with immunotherapy agents (NCT04848116, NCT03689699, NCT02451982, NCT04050462, NCT03400332, NCT04572451, and NCT04123379) and chemotherapy (NCT05148234) [193].
FLT3L
FMS-like tyrosine kinase 3 receptor ligand (FLT3L) plays an active role in the maturation of macrophage-dendritic progenitors (MDPs) into pDCs and cDCs [194]. Preclinical studies have suggested that recombinant human FLT3 ligand (rhuFLT3L) agonism can lead to an enhancement in immunologic therapies, including PD-L1 inhibition [68]. Additionally, rhuFLT3L use has been shown to aid in the abscopal effect of radiation therapy by promoting immunogenic cell death [195, 196]. A similar abscopal effect has been noted when rhuFLT3L is combined with DC vaccine therapies [197]. Finally, in PD-L1 resistant mouse models, a combination approach involving FLT3L, radiotherapy, and TLR3/CD40 stimulation promotes CD8+ T cell influx, PD-L1 responsiveness, and tumor regression both locally and in distant untreated lesions, leading researchers to focus on this combination approach for clinical trials [198].
CDX-301 is a soluble rhuFLT3L developed using Chinese hamster ovary cells, and it has been shown to be a viable, well-tolerated option for combination trials [199]. Though it provides no clinical response on its own and public-domain clinical data remain scarce, preliminary phase II data (NCT0283925) involving CDX-301 in combination with single lesion SBRT resulted in 31% of analyzed subjects (9/29) recorded partial response (PR) involving distant lesions on PET imaging 2 months following therapy, further highlighting its abscopal potential [200].
STAT3
STAT3 has been implemented in immune escape and the promotion of tumor proliferation. The immunosuppressive potential of MDSCs occurs partially due to hindrances in myeloid progenitor differentiation as activated STAT3 inhibits the expression of protein kinase C βII (PKCβII) signaling [201]. Within the tumors themselves, constitutively activated STAT3 results in increased expression of PD-L1 along with the release of immunosuppressive cytokines (IL-6, IL-10, etc.) and growth factors such as CSF-1 and VEGF [202].
Considering STAT3 contributes to both tumor growth and the promotion of tolerogenic immune cells, it is an ideal target for cancer therapy development [203]. STAT3 activation occurs following phosphorylation by Janus kinases (JAKs) and subsequent homodimerization, leading it to translocate to the nucleus and perform its transcription functions. STAT3 and JAKs are then deactivated through Src homology domain-containing tyrosine phosphatases (SHP-1/2). While certain compounds have been found to impact STAT3 phosphorylation through drug repositioning studies (celecoxib, niclosamide, and pyrimethamine) or through known JAK inhibitors (ruxolitinib and pacritinib), more selective STAT3 inhibitors have since been developed including small-molecule inhibitors (napabucasin, TTI-101, OPB-51602, OPB-31121, OPB-111077, BP-1-102, and S3I-201) and oligonucleotides (danvatirsen and STAT3 DECOY) [202, 204, 205].
While the majority of trials (NCT02753127, NCT02993731, NCT01839604, NCT00955812, NCT00657176, NCT01406574, NCT01344876, NCT01711034, NCT02178956, NCT02315534, and NCT02279719) have failed to document meaningful clinical efficacy, as monotherapy or in combination (FOLFIRI, gemcitabine, paclitaxel, sorafenib, and temozolomide), several agents that have off-target effects that lead to lower STAT3 activity are currently being explored, including SHP-1/2 agonists like SC-43 [NCT04733521] and IL-6R inhibitors like tocilizumab (NCT02767557, NCT04940299, and NCT04691817) and siltuximab (NCT04191421) [202, 206,207,208].
Strategies to functionally block immune-suppressive myeloid cellsCD47-SIRP⍺
CD47 is ubiquitously expressed on the surface of normal tissue in order to allow for immune self-recognition. This occurs when CD47 binds to SIRP⍺ which is found on macrophages and DCs [209]. Tumor cells take advantage of this system via overexpression of CD47, providing a unique immune escape mechanism that has garnered considerable interest. Within TAMs, SIRPα expression also remains high and binding to CD47 within the TME further assists TAMs in maintaining their immunosuppressive phenotype through SHP-1/2 signaling [210]. Preclinical studies have found that antagonizing CD47/SIRPα signaling results not only in augmented phagocytosis, but also in DC activation, CD8+ T cell priming, and a decrease in myeloid-driven immunosuppression through macrophage polarization and an increased M1 to M2 ratio [211, 212]. Unique inhibitors of the CD47-SIRP⍺ axis include monoclonal antibodies against CD47 (magrolimab also known as Hu5F9-G4, evorpacept, CC-90002, SRF231, letaplimab, lemzoparlimab, AO-176, TJ011133, SHR-1603, and ZL-1201), monoclonal antibodies against SIRP⍺ (BI765063, GS-0189, CC-95251), and recombinant SIRP⍺-Fc fusion proteins (TTI-621, TTI-622, and evorpacept) [213, 214]. Bispecific antibodies are also emerging with secondary targets including CD19 (TG-1801), CD20 (IMM0306), CD40L (SL-172154), PD-1 (HX009), and PD-L1 (IBI322) [215].
Developing a monoclonal antibody toward SIRPα can be challenging considering that various SIRP homologs exist alongside various SIRPα alleles, so agents require pan-allele sensitivity while avoiding SIRP homolog activity [216]. Advantages, however, include the fact that SIRP is not ubiquitously expressed, allowing for anti-SIRPα therapies to avoid the destruction of bystanders such as red blood cells, as seen with anti-CD47 agents. This also allows them to be given at lower doses while theoretically maintaining efficacy due to decreased antigen sink. Many of the monoclonal anti-CD47 agents currently developed target different epitopes and as a result, a specific subset has been found to only weakly bind to red blood cell CD47 (lemzoparlimab, magrolimab, and AO-176), allowing them to spare these cells and prevent the development of anemia [217]. Additionally, newer anti-CD47 agents have been developed with inert Fc regions (evorpacept) to further avoid this effector function, though as a result these therapies become reliant on combination therapies involving a tumor-opsonizing antibody [218]. The SIRPα-Fc fusion products are made up of IgG Fc fused to the extracellular domain of SIRPα and this structure allows for SIRPα to bind to CD47 for a longer duration by slowing clearance through the presence of the Fc domain [219]. Though affinity for native SIRPα may be lower compared to anti-CD47 mAbs, SIRPα variants have been designed to overcome this deficiency. The small molecular weights seen with these fusion proteins may also assist with their ability to penetrate TME more readily. Bispecific antibodies aim to provide dual-signaling and improve immune cell proximity, though whether this correlates to improved efficacy remains to be seen.
The most promising clinical data involve the use of magrolimab in combination with rituximab ± chemotherapy (gemcitabine and oxaliplatin) for the treatment of relapsed/refractory B cell non-Hodgkin lymphoma (NHL), where researchers noted an ORR of 50% (11/22) with CR noted in 36% (8/36) of participants (NCT02953509) [220]. Contrast this to the phase I results involving magrolimab monotherapy in the treatment of advanced solid tumors where the ORR approached 5% (NCT02216409, NCT30811285) [221]. Similarly, evorpacept in combination with pembrolizumab ± trastuzumab for advanced solid tumors (ASPEN-01 and NCT03013218) resulted in an ORR of 0% (0/15) and a disease control rate (DCR) of 26.7% (4/15) [222]. Biopsies obtained from participants post-treatment showed increases in TAM populations on immunohistochemistry staining, and no increase in CD8+ tumor-infiltrating lymphocytes (TILs) was noted in either treatment arm.
CD24-Siglec-10
CD24 suppresses inflammatory responses through binding to sialic acid-binding immunoglobulin-type lectin-10 (Siglec-10) found on the surface of macrophages [223]. However, CD24 has also recently been found to provide a unique immune escape mechanism utilized by a variety of cancer cells [224]. Though CD24 is primarily expressed on immune progenitor cells and lymphoid tissue, certain tumor types have been found to express CD24 at high magnitudes [224, 225]. To elicit an effect, CD24 binds to TAMs via surface-bound Siglec-10, resulting in immune escape through SHP-1 and SHP-2 signaling, similar to CD47. To put this theory of immune escape to the test, researchers removed the CD24 protein gene from human breast cancer cell lines, then intermixed these CD24-deficient cells with wild-type cancer cells. They confirmed that macrophages cleared out the CD24-deficient populations more rapidly [224]. These cells were also significantly more sensitive to anti-CD47 therapies, suggesting a plausible synergistic role with some of the CD47-targeting agents mentioned previously. Finally, Siglec-10 knockout macrophages were also created, resulting in improved phagocytosis abilities compared to controls.
CD24 also plays a potential role in cancer migration in various cancer types along with prognostication [226, 227]. As a result, many preclinical studies now closely evaluate targeting this signaling pathway as a way of combating both malignancies and the TME. Initial models involved unconjugated monoclonal antibodies targeting the leucine–alanine–proline (LAP) epitope of CD24 (SWA11) which led to antibody-dependent cellular cytotoxicity (ADCC) in lung, ovary, bladder, myeloma, and lymphoma models, all while notably altering the cytokine milieu and hindering metastatic potential [228]. Bispecific antibodies involving MHC-I (cG7-MICA) and CD30 have also been examined with similar results reported. Success has also been noted with antibody–drug conjugates involving various payloads including nitric oxide, pseudomonas exotoxin, and even ricin A-chain immunotoxin [228,229,230,231]. More recently, anti-CD24 chimeric antigen receptor (CAR) T cells and NK products have been investigated in pancreatic and ovarian cancer models with the use of CARs derived from SWA11, with dual targeting seeming to help reduce the incidence of off-target events [228, 232, 233].
A humanized, affinity-matured version of anti-CD24 has already been developed (ONC-781) and this monoclonal antibody has been used to construct an antibody–drug conjugate (ONC-784), a bispecific antibody to CD3 (ONC-783), and a CAR-T therapy (ONC-782) for potential clinical trials [234]. Little remains publicly available regarding clinical trial prospects, but it seems fair to say that dual inhibition of immune escape mechanisms (PD-L1, CD47, and CD28) will likely be on the horizon.
Strategies to reprogram myeloid cells to acquire pro-inflammatory propertiesTLR agonists
Sensing of DAMPs and PAMPs through TLRs expressed by APCs results in their activation and subsequent T cell priming [235]. TLR agonists are studied as adjunct therapies to tumor vaccines and immunotherapy agents to amplify treatment response. However, modifications of TLR agonists are required for clinical use to adjust for their short half-life, poor localization, and limited immunogenicity [236]. For the purpose of this review, we will be discussing TLR9 agonists which have been the most extensively studied TLR agonists within the clinical trial setting.
TLR9 is constitutively expressed within the endosomes of B Cells and pDCs, though additional myeloid subtypes have been found to express TLR9 when activated by immune triggers including infection [235]. TLR9 recognizes unmethylated cytosine-phosphate guanine (CpG) oligodeoxynucleotides (ODNs) found on modified or foreign DNA, resulting in robust activation of innate and adaptive immune cells through MyD88 signaling [237]. This discovery has led researchers to engineer TLR9 agonists based on CpG ODNs. Given these agonists are physiologic derivatives, they naturally carry shorter half-lives, but with modifications including a nuclease-resistant phosphonothioate backbone (CPG 7909, ISS 1018, CpG-28, IMO-2055, tilsotolimod, SD-101, GNKG168, and S-540956), the half-life of these agents has been increased from minutes to days [237]. Other modifications include the creation of double stem-loop immunomodulators (dSLIMs) which are CpG DNA molecules that have been covalently closed, forming a dumbbell-like shape that is resistant to DNase degradation (lefitolimod and EnanDIM) [238]. Additionally, various delivery vehicles have been explored to improve localization and bioavailability including nanoparticles (cavrotolimod) and viral-like particles (CMP-001 and NZ-TLR9) [238]. TLR9 agonists are also being investigated as conjugate payloads as part of antibody–drug conjugates for monoclonal antibodies, including anti-SIRPα (ALTA-002) and anti-CD22 (TAC-001). These have been collectively termed as “Toll-like receptor agonist antibody conjugates” (TRAAC) [239].
Preclinical data involving modified CpG ODNs in murine models have demonstrated that intratumoral injections result in tumor regression along with tumor-specific T cell responses and upregulation of immune checkpoint genes including PD-L1, OX40, and CTLA4 [240]. This has been a key justification for combining checkpoint inhibitors with CpG ODNs. Additionally, CpG ODNs are radiosensitizers in early lung cancer models with a sensitivity enhancement ratio (SER) of 1.28, further justifying a multi-therapy approach [241]. In the clinical setting, single-arm phase II results involving intratumoral injections of a CpG agonist (PF-3512676) plus local radiation in low-grade B cell lymphoma noted an ORR of 23.3% (7/30) with a DCR of 86.6% (26/30) [242]. Ongoing phase I studies are examining the use of CpG ODNs in combination with local radiation and immunotherapy agents for the management of refractory lymphomas (NCT03410901).
PF-3512676 (CPG 7909) is the most extensively studied clinical CpG ODN, particularly in combination with conventional chemotherapy (paclitaxel, carboplatin) for the treatment of NSCLC. Initial phase II trials appeared promising with improvements in OS compared to chemotherapy alone but following the release of interim results from two phase III trials, both trials were terminated due to high rates of sepsis-related events and minimal evidence of improved clinical efficacy [243]. CpG ODNs continue to be studied as adjuncts, particularly in the realm of cancer vaccine therapies given their immunostimulatory properties.
CD40 agonists
CD40 is readily expressed on antigen-presenting cells and is essential to their activation. Additionally, its ligand CD40L is found on a variety of immune and non-immune cells, including CD4+ T cells. CD40L helps with the cross-priming of CD4+ cells to non-self-antigens by providing a co-stimulatory effect [244]. Activation of CD40 on DCs leads to upregulation of MHC molecules, increase in IL-12 secretion, and the promotion of cytotoxic T cell activation [




留言 (0)