
記住我
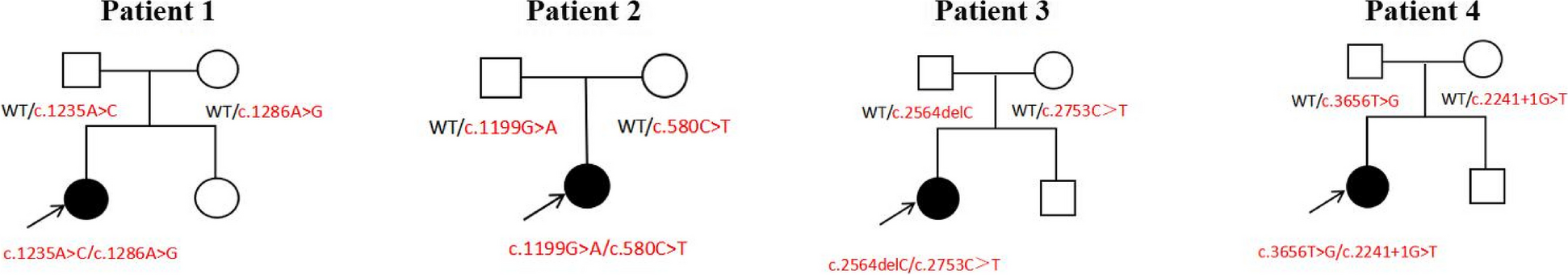
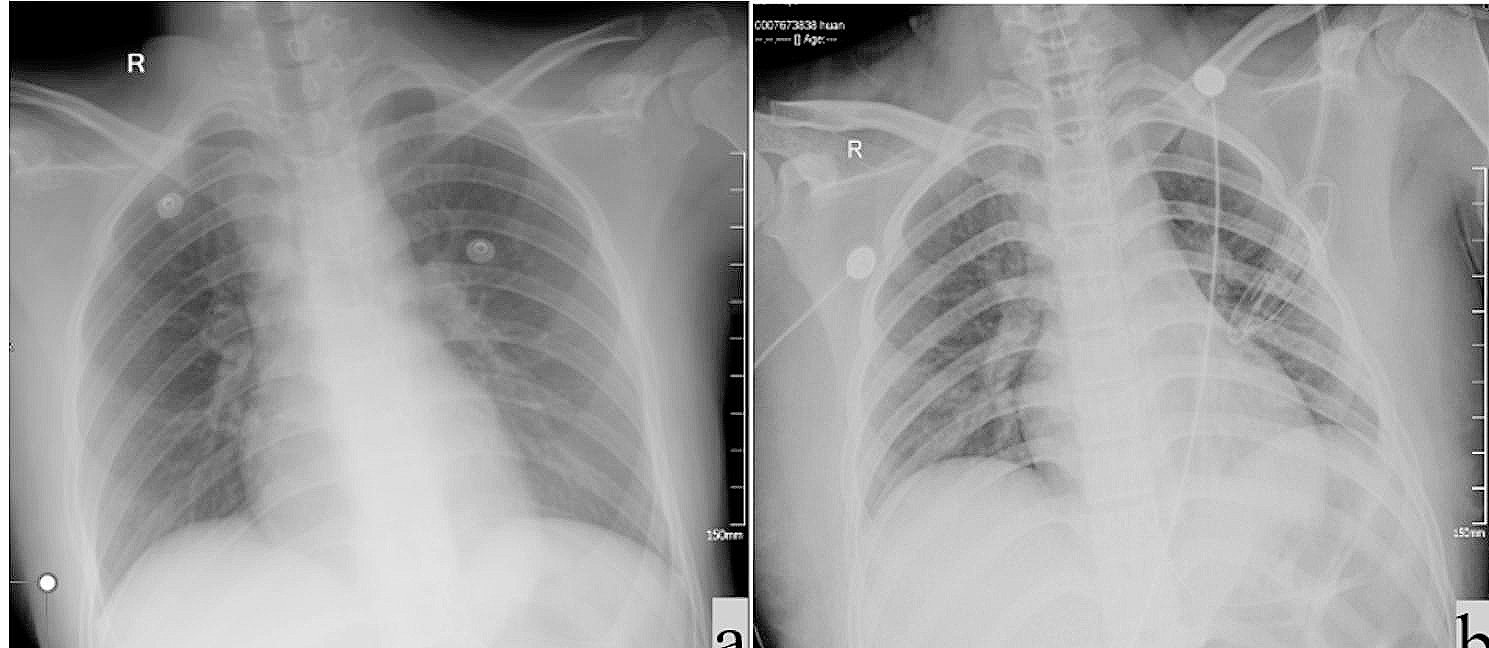



A male newborn was delivered at 31+4 weeks of gestation (WG), due to preterm labor, by spontaneous vaginal birth. The parents were healthy and non consanguineous. Family history disclosed a brother, born prematurely at 28 WG and died for sepsis on the twelfth day of life, and a sister born prematurely as well, at 31 WG, and affected with an unspecified congenital skin disorder. Our patient had two further brothers, reported as healthy. Pregnancy was not followed either by obstetrics or other health professionals, and clinical data were thus not available. On day 2 of life he was transferred to our Department. At admission, physical examination showed large skin lesions with hyperkeratotic plaques and diffuse hemorrhagic areas, necrosis of the fingers of both hands, generalized subcutaneous edema, flattened dysplastic ears, ectropion, eclabium, macrostomia and macroglossia. Pseudocontractures of the limbs (especially of fingers and toes), resulting from cutaneous constriction, were also observed (Fig. 1). Spontaneous motility and reactivity were markedly reduced, while axial muscular tone was increased. Head and abdominal ultrasound (US) documented no abnormalities. Conversely, echocardiography showed pulmonary hypertension (pulmonic artery pressure around 35–40 mmHg), along with patent foramen ovale. Due to severe ectropion, ophthalmological evaluation was not carried out. Target NGS of 27 genes associated to congenital ichthyosis was performed, and identified the compound heterozygous mutations c.233_234del (p.Thr78ArgfsTer3) and c.1287 + 2_1287 + 5del of the ABCA12 gene. Such variants were inherited from the mother and the father respectively, and them both are not reported in literature. They were then tested with Sanger sequencing, confirming the HI diagnosis (which was however available about ten days after NGS analysis was carried out), also according to the phenotype of the newborn. Postnatally, he was supported by invasive mechanical ventilation and total parenteral nutrition and later, from day 5, by minimal enteral feeding. During the first days of life blood evaluations were minimized. Laboratory tests documented hypernatremic dehydration (plasmatic sodium 153 mEq/l, chloride 122 mEq/l, and osmolarity 297 mOsm/l), which required rehydration through central venous catheter. Meanwhile, due to diffuse subcutaneous edema related to hypoproteinemia along with hypoalbuminemia, infusions with albumin, protein solution and plasma were administered. On day 6, owing to the relevant difficulties in finding venous accesses and to obtain blood samples, a femoral vein catheter was surgically positioned. In the following days, due to the worsening of hard compressive plaques on chest, forearms and legs, and based on a multidisciplinary assessment (neonatologist, pediatric surgeon, plastic surgeon, anesthesiologist and clinical geneticist), an escarotomy intervention was performed in the NICU. The following clinical course was marked by anemia, which required red blood cells transfusion, in addition to a sepsis, sustained by Escherichia Coli and treated with aminoglycoside antibiotic therapy. On the tenth day of life, a progressive decay of the general conditions, mainly linked to the worsening of the respiratory distress secondary to restrictive pulmonary disease, occurred. This led to lung failure, unresponsive to maximal mechanical ventilation and inotropic drugs, and finally to death.
Fig. 1
Patient 1. Hyperkeratotic plaques and diffuse hemorrhagic areas, necrosis of the fingers of both hands, generalized subcutaneous edema, pseudocontractures of the limbs (especially of fingers and toes)
Patient 2A female newborn, delivered at 39+1 WG by spontaneous vaginal birth, was the second child of healthy and non consanguineous parents. Family history was negative for genetic diseases and/or genodermatoses, and also pregnancy was uneventful. On the first day of life, blisters with yellow content appeared in the lower anterior surface of the abdominal wall, pubic and perianal region, proximal segments of both thighs, and inguinal areas (Fig. 2A). Therefore, she was admitted to the Neonatology Unit. Within 48 h, the newborn developed fever, associated to generalized erythematous skin lesions with progressive bullous eruptions, in addition to further superficial blisters breaking under slight pressure. The Nikolski sign (easy separation of skin layers under application of horizontal, tangential pressure to the skin) was present. Mucosal surfaces were intact. Inspection of external genitalia showed de-epithelizing lesions in the inner and outer vulvar labia. Clinical and US examination of other organs and systems revealed no abnormalities. Laboratory tests showed neutrophilic leukocytosis and increased C-reactive protein, leading to the diagnostic hypothesis of a bacterial infection. In the suspicion of a staphylococcal scalded skin syndrome (SSSS), intravenous (iv) antibiotic therapy with oxacillin and gentamicin (this latter also topically applied) was empirically begun, and then changed into iv vancomycin, based on the results of skin swab culture and antibiogram (growth of an oxacillin-resistant coagulase-negative staphylococcus hominis). Erythroderma and the other skin lesions decreased in 10 days, and the patient was discharged on day 15, and included in a multidisciplinary (auxological and dermatological) follow-up. At age 1 month, new blisters in the gluteal region were observed, in addition to re-epithelization of the previously described lesions. At 2 months of age, after the use of protective padding, bullous lesions decreased. However, minimal desquamation, with small areas of erythematous skin erosions (seen also on dermatoscopy), were still present in the diaper region (Fig. 2B). A new skin culture, as well as blood tryptase analysis and zinc assay were performed and resulted negative, ruling out thus re-infection, mastocytosis and enteropathic acrodermatitis. Then, due to persistence of the skin lesions, it was decided to proceed in the diagnostic work-up. In the suspicion of congenital epidermolytic ichtyosis (EI) or epidermolysis bullosa (EB), target NGS analysis of the genes involved in both cutaneous diseases was performed in the patient and her parents. Genetic investigation revealed in the baby the heterozygous de novo variant c.1327A > G of the KRT1 gene, causing the amino acid change p.Lys443Glu at the protein level, for EI diagnosis. Topical treatment with hydrating, emollient and re-lipidant creams, along with detergent oil for daily hygiene (already started after the evaluation at 1 month of age) was continued, and fusidic acid cream was added in the residual erythematous bullous lesions. The following clinical course was characterized by few minimally de-epithelizing lesions on trunk and skin folds, without other cutaneous alterations. She actually is 9 months old, and shows regular growth and development. Her general clinical conditions are good, and the skin picture is outlined by mild hyperkeratosis on the anterior surface of the neck (Fig. 2C), and on the axillary and palm-plantar regions, associated to minimal persistent bullous lesions in the diaper region. In addition, the mother reports occasional pungent odors from the hands. The previously administered topical treatment is currently continued, along with alternating use of a mild antiseptic detergent.
Fig. 2
Patient 2. A Blisters with purulent content on lower anterior surface of the abdominal wall, pubic region, proximal segments of both thighs, and inguinal areas. B Small areas of erythematous skin erosions seen on dermatoscopy. C Mild hyperkeratosis of the anterior surface of the neck
Patient 3A male newborn was delivered at 40+6 WG, by cesarean section for fetal bradycardia. Parents were healthy and consanguineous (second cousins). Family history was negative for genetic diseases and/or genodermatoses, and disclosed five previous miscarriages. Current pregnancy was uneventful. At birth, physical examination showed diffuse thick skin, with xerosis and wide hyperkeratotic areas affecting scalp, face, ears, neck, chest and lower back. No collodion membrane was observed. Sparse scalp hair (with receding front attachment) and eyebrows, cutaneous hyperlinearity of the forehead, absent eyelashes and blepharophimosis, in addition to dysplastic ears with thick helices outlined the craniofacial profile. Fissured wrists, axillary regions and ankles, were also noted (Fig. 3A/B/C/D). Follicular atrophoderma, palmoplantar keratoderma, nail and mucosal changes were not observed, and sweating was normal. He was then conducted to the Neonatology Unit, to start the diagnostic work-up. Laboratory tests (including T-lymphocytes subsets and total immunoglobulin levels) and multiorgan US evaluations showed normal findings. Auditory brainstem response (ABR) documented no abnormalities, while ophthalmological assessment disclosed blepharitis. In the suspicion of congenital ichthyosis, target NGS analysis of the main associated genes found the homozygous variant c.1231G > T of ST14. The identification of its mutation, which leads to the introduction of the premature stop codon p.Gly411Ter, confirmed the ARIH diagnosis. Genomic analysis was then extended to parents, who were found to be heterozygous carriers of the same nonsense variant. Such mutation is not present in the database of allele frequencies (Genome Aggregation Database, gnomAD), nor it is reported in literature. The following clinical course occurred without complications. He was soon positioned into the baby incubator, with increased levels of temperature and humidity. On day 3, he was moved to the open cradle, and the clinical and laboratory monitoring for the possible appearance of fever and/or other signs of inflammation, identified no abnormalities. In the meantime, he started daily hygiene treatment with detergent oil, topical therapy with emollient/re-epithelizing creams applied on the fissured skin regions, along with gentamicin cream on those with erosion. Cleansing drops were administered in the ears, while lubricant solutions based on 0.2% sodium hyaluronate were used for the eyes. He was discharged at around 1 week in good general conditions, and included in a multidisciplinary (dermatological, auxological, ophthalmological, immunological) follow-up. At age 1 month, complete desquamation of trunk and lower region of the face were observed. Specifically, scabs with underlying erythematous skin in the frontal and periocular regions, associated to scaly plugs into the external ear canal were noted, and for which a topical treatment with 0.1% mometasone furoate cream (1 mg/g) was added. At age 4 months, the cutaneous alterations progressively improved, especially those of the face, which appeared smooth and without scales. Crusts on scalp (associated to erythematous skin compatible with seborrheic dermatitis), sacral region and lower limbs were, however, still observed. Therefore, he underwent exclusive treatment with emollient and hydrating creams, in addition to the application in the scalp of 0.1% mometasone furoate ointment. He currently is 9 months old, and shows normal growth and neuromotor development. The skin picture is outlined by remission of the previous lesions, except for minimal scaling of the scalp. Hair (Fig. 4A/B), eyelashes and eyebrows are less, but still sparse. In addition, marked photophobia, which makes him unable to keep the eyes open outdoors, followed by immediate tearing, is reported.
Fig. 3
Patient 3. Diffuse thick skin, with xerosis and wide hyperkeratotic areas. A Sparse scalp hair (with receding front attachment) and eyebrows, cutaneous hyperlinearity of the forehead. B Absent eyelashes and blepharophimosis. C Dysplastic ear with thick helix. D Fissured wrist and axillary region
Fig. 4
Patient 3 at age 9 months: less marked sparse hair
留言 (0)