
記住我
With the human genome being almost complete, the estimated number of protein-coding genes has stabilized at around 20,000 (Lander et al., 2001; Pertea et al., 2018). However, there is nearly six times the number of annotated proteins (115,885 in Ensembl GRCh38.p13, September 2021). The key to this diversity lies in the fact that every step in the production and processing of messenger RNA (mRNA), from transcription to translation, provides an opportunity for molecular diversification. Among all mRNA processing steps, perhaps the most well-studied mechanism and possibly the one that provides the greatest chance for molecular variation in eukaryotes is splicing. First described in 1977, splicing is a sequential set of biochemical reactions to remove introns from the pre-mRNA molecule (Berget et al., 1977; Chow et al., 1977). The discovery of splicing came with the observation that the same adenoviral gene produced different transcripts (Berget et al., 1977; Chow et al., 1977). The modular structure of the pre-mRNA, with exons and introns, facilitates the assembly of different configurations of exons to generate alternative transcript isoforms (Gilbert, 1978), that is, alternative splicing. Soon after the discovery of splicing, the first case of alternative splicing was described in humans in the immunoglobulin μ gene, where one of the isoforms encoded the membrane-bound antibody. In contrast, the other isoform encoded the secreted form of the protein (Singer et al., 1980). In recent decades, high-throughput sequencing techniques have provided evidence indicating that more than 95% of the genes may undergo alternative splicing (Barbosa-Morais et al., 2012; Pan et al., 2008; Wang et al., 2008). These results have highlighted the vast impact that alternative splicing has on shaping the transcriptome. However, it remains unclear how frequently transcriptome variation results in measurable protein and functional changes.
A shift in the relative abundances of the transcript isoforms produced from a gene locus may lead to the production of protein variants, which may in turn impact gene function through various mechanisms, including the alteration of protein–protein interactions (Ellis et al., 2012; Weatheritt et al., 2012; Wojtowicz et al., 2007). However, in vivo studies using cutting-edge proteomics have failed so far to validate these functional impacts, sparking an intense debate on the field (Blencowe, 2017; Tress et al., 2017). In addition to the intrinsic differences and limitations of RNA sequencing and proteomics technologies, multiple regulatory mechanisms controlling transcript stability and translation could hinder the identification of direct links between transcript and protein variation (Braun & Young, 2014). Despite the abundant evidence supporting the functional impact of alternative splicing (Baralle & Giudice, 2017), whether it leads to fundamentally distinct proteins remains unknown for most genes. Understanding the regulation and interdependency of splicing with other regulatory processes is crucial to explain how the transcriptome encodes the functional complexity of cells. Moreover, to fill in the knowledge gaps, from gene expression to the production of different functional proteins, it is essential to gain insight into the mechanisms of RNA processing in the context of both the transcriptome and the translatome. In this review, we describe the mechanisms by which changes in RNA processing may result in the translation of different protein products, the high-throughput techniques that have been developed to uncover these mechanisms, and the results from recent studies regarding the impact of alternative splicing in protein production and gene function.
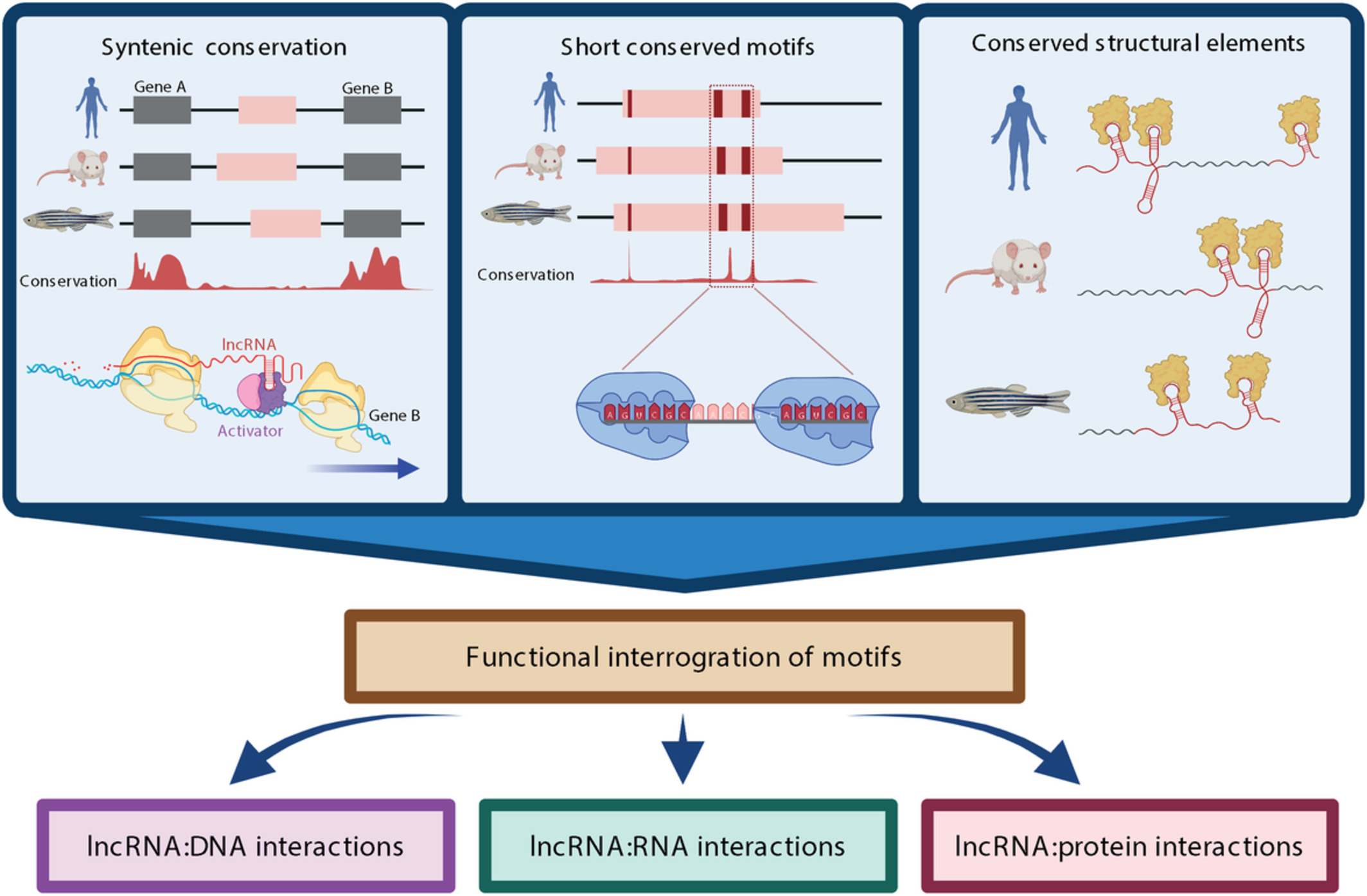
2 MECHANISMS OF TRANSCRIPT DIVERSITYThe splicing reaction starts with the recognition of sequence elements in the precursor messenger RNA (pre-mRNA) by a small nuclear ribonucleoprotein (snRNP) complex, the spliceosome, which acquires its catalytic activity while assembling on the pre-mRNA (Zhan et al., 2018; Figure 1a). In this process, intron sequences are excised from the pre-mRNA, and exon sequences are spliced together to form the mRNA. Several sequence elements in the pre-mRNA are essential for splicing. Most introns are characterized by the nucleotides GU at the 5′ splice site (SS) and AG at the 3′ SS, with very few exceptions (Figure 1a; Thanaraj & Clark, 2001). The branch point (BP) consists of an A nucleotide with a catalytic role near the 3’SS, and a polypyrimidine tract (PPT) is located between the BP and the 3′ SS. However, BPs may also occur far upstream of the 3′SS (Corvelo et al., 2010). The BP and PPT show specific sequence motif biases, but these may vary between species (Schwartz et al., 2008). The core spliceosome comprises five main snRNPs (U1, U2, U4, U5, and U6) that are sequentially assembled in the mRNA molecule, where U1 recognizes the 5′ SS and U2 the BP. Several conformational and compositional rearrangements involving U2, U5, and U6 take place to allow two transesterifications: the first one between the 5′ SS and the BP and the second between the 5′ end of the first exon and the 3′ end of the second exon (Figure 1a).

The mechanisms of splicing and alternative splicing. (a) The complexes and processing steps of splicing for the major spliceosome are depicted. In the assembly phase, U1, U2, U4, U5, and U6 are sequentially assembled in the mRNA molecule resulting in the formation of complex E, complex A, and finally, complex B. The activation phase consists of conformational and compositional rearrangements of complex B involving U2, U5, and U6. Two transesterifications occur as the catalytic complex C adapts its conformation: the first between the 5′ SS and the BP and the second between the 5′ end of the first exon and the 3′ end of the second exon. Finally, the snRNPs still attached to the mRNA disassemble to start a new cycle. (b) Illustration of the regulation of splicing by cis- and trans-acting factors. Cis-acting factors are represented as boxes in introns and exons. Trans-acting factors are represented as circles bound to their corresponding cis-acting elements: exonic splicing enhancer (ESE), exonic splicing silencer (ESS), intronic splicing enhancer (ISE), or intronic splicing silencer (ISS). Silencers are depicted in orange and enhancers in light blue. (c) Schematic representation of alternative splicing events. Gray boxes represent constitutive exons, red and orange boxes represent alternative exons, and a light blue box represents an exonized intron. Discontinuous ends in the exons indicate that the transcript continues in that direction. Thick blue lines represent introns in the transcript before processing, and dashed lines above and below represent the alternative processing of the exons
2.1 Alternative splicing regulationExon recognition is not straightforward, especially in metazoans, considering that the sequence surrounding the splice sites and the branch point can be highly variable, and introns are on average thousands of times larger than exons (Iwata & Gotoh, 2011; Lander et al., 2001; Long & Deutsch, 1999). For this reason, for efficient splicing to occur, additional cis-acting elements on the pre-mRNA sequence and trans-acting proteins are often required (Figure 1b; Lim & Burge, 2001). These additional regulatory elements, which work together with the core splicing signals (3′SS, 5′SS, and BP), are not just essential for catalysis but often act as modulators of the splicing process. They are classified according to their location on intron or exons and their role in either enhancing or repressing splicing, resulting in four main classes: exonic splicing enhancer (ESE) or silencer (ESS), and intronic splicing enhancer (ISE) or silencer (ISS; Wang & Burge, 2008). Whereas most of these regulatory elements are located near the splice sites, distant ones have also been described to modulate exon inclusion (Lovci et al., 2013). Generally, these sequence motifs are short and represent binding sites for RNA binding proteins (RBPs; Fu & Ares, 2014). These RBPs are the trans-acting factors that regulate exon definition and recognize the cis-acting elements with variable affinity and specificity.
Two prominent RBP families that regulate splicing are SR proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs), which often display opposite regulatory roles. SR proteins have one or two RNA-recognition motifs and arginine/serine (RS)-rich domains that mediate protein–protein interactions (Jeong, 2017). They generally bind ESEs to enhance exon inclusion but can exceptionally act as inhibitors by binding ISSs close to the intron boundaries (Bradley et al., 2015; Shen & Mattox, 2012). On the other hand, hnRNPs also show a modular structure with multiple RNA binding and auxiliary domains (Geuens et al., 2016). They bind ESSs to repress splicing, but as SR proteins, they can have an enhancing role depending on their position relative to other sequence elements, for example, by binding ISEs (Erkelenz et al., 2013). Multiple other RBPs have been found to play a role in splicing regulation in different physiological and disease contexts (Yee et al., 2019).
Numerous efforts have attempted to provide a complete census of proteins involved in splicing regulation, uncover their cognate sequence motifs, and establish their role in splicing, which have been jointly dubbed as the splicing code (Barash et al., 2010). These have been primarily based on cross-linking and immunoprecipitation (CLIP) followed by high-throughput sequencing, and have allowed identifying many RBPs and their recognition motifs, as well as other splicing regulators including coactivators or proteins involved in other RNA-processing steps (König et al., 2011; Ule et al., 2003; Van Nostrand et al., 2020). However, the splicing code remains challenging to decipher due to the many proteins and sequence elements contributing to the splicing outcome. First, the strength of the splice sites, measured as their similarity to the consensus, dictates the susceptibility to regulation by trans-acting factors. Strong splice-site motifs facilitate splice-site recognition, but weaker splice sites are more favorable for effective splicing modulation (Baralle & Baralle, 2018). Second, it is still challenging to determine if the binding of an RBP will positively or negatively regulate splicing since many of them have dual roles depending on the context (Fu & Ares, 2014). Finally, and most importantly, splicing patterns are driven by complex combinations of proteins and sequence motifs with cooperative and competitive relationships (Papasaikas et al., 2015; Ule & Blencowe, 2019).
2.2 Variation of the splicing productsThe variation in splice site selection is generally described as local variations, which can be categorized into five primary events (Figure 1c; Box ). The selection of alternative splice sites can make exons shorter or longer, generating alternative 5′ SS (A5) and alternative 3′SS (A3) splicing events. Splicing variation can also affect complete exons that may be spliced in or out of the mature transcript, described as a skipping exon (SE) event. Mutually exclusive exons (ME) are a complex case where one of two adjacent exons may be skipped, and they both are rarely observed together in a mature transcript. Finally, intron retention (IR) occurs when an intron is included in the mature transcript. Different combinations of primary events increase the number of isoforms produced from a single gene changing the UTRs or the coding sequence. Exons are classified as alternatives when they are differentially included in isoforms from the same gene, possibly described according to the event types mentioned above, or constitutive when present in all the transcript variants. On the other hand, these alternative splicing events coexist with other forms of transcript variation. The choice of alternative promoters can induce the selection of alternative first (AF) exons, and alternative polyadenylation (APA) can lead to the inclusion of alternative last (AL) exons (Figure 1c). Due to the involvement of regulatory RBPs that are common to splicing regulation, AF and AL events are often included when studying alternative splicing.
One or more alternative splicing events can give rise to a wide range of transcript-isoform outputs, from two to a few hundred per gene (Ray et al., 2020; Tung et al., 2020), leading to complex patterns of coordinate splicing events (Grosso et al., 2008; Licatalosi & Darnell, 2010; Tapial et al., 2017). How they exactly contribute to functional diversification is still an active area of research. Complex alternative splicing patterns have been described to contribute to the definition of embryonic stem cells, to cell lineage differentiation processes, or to the epithelial–mesenchymal transition (Agosto & Lynch, 2018; Kalsotra & Cooper, 2011; Venables et al., 2013). Differentiated cells and tissues also show specific splicing patterns, as it has been described for the brain, heart, skeletal muscle, testis, and liver, as well as for erythrocytes and immune cells, among others (Baralle & Giudice, 2017; Bhate et al., 2015; Licatalosi & Darnell, 2010; Lynch, 2004; Nakka et al., 2018; Pimentel et al., 2016; Tapial et al., 2017). Finally, alternative splicing is also related to physiologic response to changing conditions, such as DNA damage response, thermal regulation, or even adaptation to stress conditions (Biran et al., 2020; Haltenhof et al., 2020; Shkreta & Chabot, 2015).
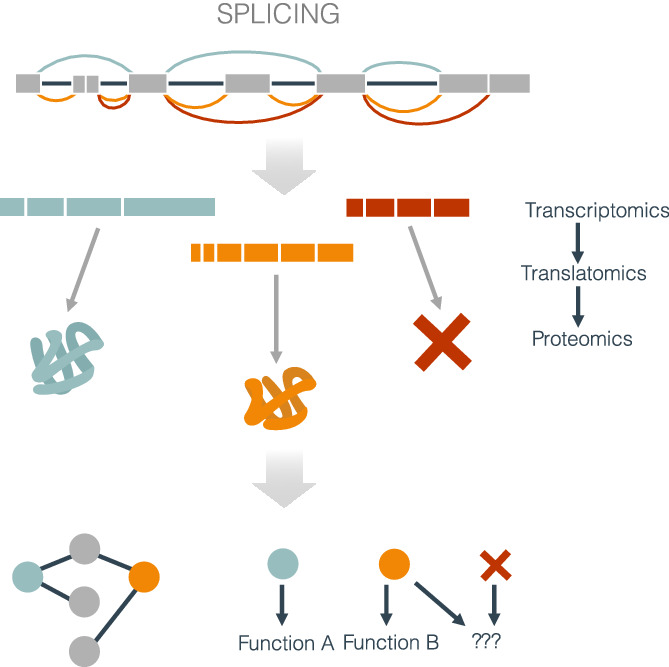
2.3 Interplay of splicing with other RNA processing stepsDuring transcription initiation, expression of different transcription factors (TFs) and changes in chromatin state can lead to the selection of alternative promoters, thereby triggering transcription from an alternative transcription start site (Davuluri et al., 2008). As a result, different exons may be included at the 5′ end of the transcript that can modify the N termini of the protein (Leppek et al., 2018). This may happen as a direct sequence change of the open reading frame (ORF) of the resulting transcript (Tasic et al., 2002) or through a change in the translation start site mediated by alterations in the regulatory sequences of the 5′ untranslated region (5′UTR) of the mRNA (Figure 2). On the other hand, there is often no observable effect in the protein product. Instead, changes in the 5′UTR may affect the translation efficiency, that is, two or more of the transcript isoforms produced from the gene locus encode the same protein but at a different rate. (Tamarkin-Ben-Harush et al., 2017; Figure 2).
 How alternative splicing potentially reshapes the protein products. Alternative splicing (AS) can reshape the proteome in diverse ways. These can be separated into two impacts: changing the protein sequence (upper panels) or altering the amount of protein produced (lower panels). Splicing changes affecting the coding sequences (CDS) can generate alternative proteins with different amino acid sequences, structures, and functions. Similarly, changes in the 5′ end of the mRNA molecule can lead to an alternative initiation site, which results in proteins with different amino-terminal ends. Both mechanisms could also lead to a shift in the reading frame. Premature stop codons generated through a splicing change can translate into truncated proteins or lead to mRNA decay (Box ). Finally, translation efficiency can be affected by the inclusion of alternative 5′UTRs through alternative splicing
How alternative splicing potentially reshapes the protein products. Alternative splicing (AS) can reshape the proteome in diverse ways. These can be separated into two impacts: changing the protein sequence (upper panels) or altering the amount of protein produced (lower panels). Splicing changes affecting the coding sequences (CDS) can generate alternative proteins with different amino acid sequences, structures, and functions. Similarly, changes in the 5′ end of the mRNA molecule can lead to an alternative initiation site, which results in proteins with different amino-terminal ends. Both mechanisms could also lead to a shift in the reading frame. Premature stop codons generated through a splicing change can translate into truncated proteins or lead to mRNA decay (Box ). Finally, translation efficiency can be affected by the inclusion of alternative 5′UTRs through alternative splicing
The coupling of splicing with transcription has been widely studied through the RNA Polymerase II (RNAPII) function. From the kinetic point of view, it has been shown that elongation rates of RNAPII can affect SS recognition with different outcomes depending on the SS strength (Fong et al., 2014; Saldi et al., 2017). On the other hand, the recruitment model is supported by known interactions of several SFs with RNAPII, mainly through the C-terminal domain (CTD) of the large subunit or with other members of the elongation complex to regulate exon inclusion (Huang et al., 2012; de la Mata & Kornblihtt, 2006; Hsin & Manley, 2012; Das et al., 2007). For example, direct binding of U2AF on the CTD has been shown to recruit Prp19, crucial for spliceosome activation (David et al., 2011). A new approach in a recent study proposes multiple interactions between U1 and RNAPII that support a model of co-transcriptional assembly of the spliceosome on the RNAPII (Zhang, Aibara, et al., 2021). Beyond RNAPII, it has also been observed that the use of alternative promoters, enhancers, and TFs affect splicing rates (Han et al., 2017; Alasoo et al., 2019).
Chromatin state has also been described as affecting the splicing outcome (Agirre et al., 2015, 2021; González-Vallinas et al., 2015). Indeed, DNA methylation and histone posttranslational modifications (PTMs) can also modulate splicing and potentially participate in alternative splicing. Alternatively, spliced exons show lower methylation levels than constitutive ones (Gelfman et al., 2013). Moreover, depending on the factors mediating the crosstalk between methylation and splicing, the effect can be either promote or repress exon inclusion (Shukla et al., 2011; Maunakea et al., 2013; Yearim et al., 2015; Brito et al., 2020). Similarly, distinct histone modification signatures are also known to correlate with specific splicing patterns. This has been observed for FGFR2, where combinations of histone methylation marks regulate two mutually exclusive isoforms (Luco et al., 2010). This and other similar cases have led to the proposal of a chromatin code that can predict alternative splicing (Agirre et al., 2015, 2021).
Alternative polyadenylation (APA) is emerging as a relevant mechanism to regulate gene expression in multiple biological processes (Ren et al., 2020). APA can result in significantly different 3′ UTRs, affecting the transcript stability, export, and translation rates (Mayr, 2016; Kishor et al., 2020). Longer 3′ UTRs generally contain more regulatory structures and are related to decreased mRNA stability and translation efficiency (Moqtaderi et al., 2018). Moreover, if APA occurs upstream of the last exon, it can affect the 3′ UTR and the coding region by changing the exon composition at the 3′ end of the transcript. APA in coding regions can regulate gene expression and contribute to protein diversification (Tian & Manley, 2016). Apart from the core 3′ end processing factors, APA can be controlled by the interplay with other RNA-processing steps, including transcription (chromatin state, TFs, and RNAPII activity) and splicing (Elkon et al., 2013; Gruber & Zavolan, 2019). RNA methylation dynamics have also been shown to regulate APA usage, promoting inclusion of terminal exons and therefore controlling the 3′ UTR length (Bartosovic et al., 2017).
Splicing regulators also influence other RNA biology processes that can alter the transcriptome beyond alternative splicing itself. For instance, members of the SR and hnRNP protein families can regulate transcription, processing, stability, and transport (Huang & Steitz, 2005; Ji et al., 2013; Geuens et al., 2016).
Furthermore, translation can be regulated to modulate the amount of protein produced and generate alternative proteins by the alternative selection of Translation Initiation Sites (TISs). Alternative TISs, often downstream of the canonical one, can be selected through several mechanisms, including leaky scanning of the ribosome, presence of non-AUG codons, or through uORFs affecting the capacity of the ribosome to reinitiate translation (Kozak, 2005; Kochetov, 2008). In other cases, the start site of an uORF becomes the actual start site of a 5′ extended version of the translated ORF (Kochetov, 2008). In addition to the variation in the N-termini of the protein, alternative TISs can also shift the translating frame, potentially resulting in different protein products and often creating premature stop codons (Orr et al., 2020).
Finally, biochemical RNA modifications are emerging as a mechanism to modulate the stability and translation efficiency of mRNAs (Boo & Kim, 2020). RNA modifications are abundant in ribosomal (rRNA), and transfer RNAs (tRNA), and their alterations impact translation through the functional impairment of rRNAs and tRNAs (Liu, Clark, et al., 2016; Ma, Wang, et al., 2019). There is now evidence that RNA modifications are also abundant in mRNAs (Dominissini et al., 2012; Squires et al., 2012), and the modifications 5-methylcytidine and N4-acetylcytidine have been linked to enhanced translation efficiency (Arango et al., 2018; Schumann et al., 2020; Huang et al., 2019). Given that there are more than 100 possible modifications (Linder & Jaffrey, 2019), this opens up multiple new possible mechanisms to modulate the protein output from mRNAs.
3 TOWARD IDENTIFYING THE EFFECTS OF SPLICING ON PROTEIN DIVERSITYTranscriptomics studies have provided insight into how AS gives rise to transcript diversity in the cells. However, given the plethora of mechanisms to control and regulate protein synthesis after pre-mRNA splicing (Box ), the transcriptome is not a trustworthy representation of the proteome. Then, the key to determining the functional impact of AS is to understand how splicing events reshape the protein products but also to discriminate which alternative transcript isoforms will be translated into protein.
Most genes have one transcript that encodes the most ubiquitously expressed and evolutionary conserved protein isoform, and best represents that gene's function. This has motivated the definition of principal transcripts, which simplifies the transcriptome based on protein sequence, structure, function, and conservation (Rodriguez et al., 2013, 2018). However, this definition clashes with evidence indicating that the most expressed transcript isoform from a gene may vary between tissues and disease conditions (Gonzàlez-Porta et al., 2013; Sebestyén et al., 2015). Changes in the most expressed protein-coding transcript isoform can thus result in a diversification of proteins and their function, leading to changes in catalytic activities, interactions with other proteins, subcellular localization, and loss of function (Yang et al., 2016; Figure 2).
3.1 Proteomics evidence of alternative splicingOne of the primary sources of evidence to support the production of multiple proteins from the same gene locus through AS is mass spectrometry (MS; Blakeley et al., 2010). In a general MS workflow, proteins are digested with enzymes, and the generated peptides are ionized and loaded into a mass spectrometer. The generated spectra are used to computationally reconstruct peptide sequences using a database of annotated proteins as guide (Aebersold & Mann, 2003; Deutsch et al., 2008). Even though the existence of alternatively spliced isoforms from RNA-seq analysis is incontestable, MS experiments have offered very little support for alternative protein products associated with AS, raising controversy in the field (Tress et al., 2017; Blencowe, 2017).
Although the detection power may vary significantly depending on the annotation database used, several studies have provided evidence for hundreds to about one thousand detectable alternative protein isoforms, often showing a tissue-specific pattern (Wilhelm et al., 2014; Kim et al., 2014; Tay et al., 2015). A study of 30 human samples identified proteins corresponding to 84% of the total annotated protein-coding genes and mapped peptides uniquely to approximately 400 alternative protein isoforms (Kim et al., 2014). In contrast, analysis of eight large-scale human proteomics datasets with highly conservative filters yielded evidence for about 200 alternative protein isoforms (Ezkurdia et al., 2015; Abascal et al., 2015). It has also been claimed that there is an overestimation of alternative protein isoforms resulting from considering cases where peptides only map to one side of the variable exon in a splicing event rather than both sides, and from not taking into account the potential false positives in the MS data processing (Tress et al., 2017).
Although a significant amount of high-throughput MS data have been produced to date for multiple cell models and tissues, studies using this data to search for protein isoform variants have almost invariably been performed in steady-state conditions (Alfaro et al., 2017; Nusinow et al., 2020). This contrasts with the fact that most RNA splicing changes have been reported in association with changing physiological conditions or between normal and disease states. Supporting this notion, several studies have provided evidence for several genes with detectable protein isoform variation across the cell cycle in leukemia cells (Ly et al., 2014).
Integration of RNA-seq experiments with MS for the identification of protein isoforms, also known as proteogenomics, has been implemented to reduce peptide mapping uncertainty and to detect novel peptides (Jeong et al., 2018; Liu et al., 2017; Wu et al., 2019; Lau et al., 2019; Agosto et al., 2019). In the context of depletion of a spliceosome component and using quantitative proteomics, it was shown that out of 450 transcripts with significant changes in RNA relative abundance, up to 160 showed consistent changes in protein production using quantitative proteomics (Liu et al., 2017). Similarly, a splice-junction-centric approach applied to public proteogenomics data led to the identification of 1500 alternative protein isoforms (Lau et al., 2019). Most of the junctions (~60%) arose from SE events, consistent with what has been observed in RNA-seq experiments.
Proteogenomics studies have raised the issue of the poor correlation between transcript and protein abundances, which is usually below 0.5 (Pearson correlation coefficient) and varies across tissues (Kosti et al., 2016; Eraslan et al., 2019; Liu et al., 2017). Despite the general lack of consistency between transcript and protein abundances, it has been observed that above certain levels of transcript expression, RNA-seq is a good predictor of protein expression (Vogel & Marcotte, 2012). A recent study has modeled the effect of protein synthesis regulation after pre-mRNA splicing and has managed to explain 30% of the variance in the protein-to-mRNA ratios (Eraslan et al., 2019).
Many of the studies mentioned above point out that spectra generated in MS experiments might not provide enough resolution to distinguish particular isoforms (Tay et al., 2015; Liu et al., 2017; Kosti et al., 2016; Eraslan et al., 2019). On the one hand, these limitations could be attributed to the digestion enzymes used in proteomics. Trypsin is the standard protease in shotgun proteomics. It efficiently digests at K or R residues, resulting in small size (<3 kDa) peptides with a positive charge at the C-terminus, which is optimal for the MS protocols (Huang et al., 2005; Laskay et al., 2013). The main drawbacks of the peptides produced are the short lengths (around six aa) and the limited proportion of the covered proteome (Giansanti et al., 2016). Other proteases such as chymotrypsin, LysC, LysN, AspN, GluC, ArgC, elastase, and proteinase K have been shown to cover complementary fractions of the proteome, and proteases such as GluC improve the detection of peptides containing PTMs, which are otherwise overlooked (Janssen et al., 2019); suggesting that combinations of enzymes may provide the best approach to obtain a comprehensive peptide identification (Dau et al., 2020; Giansanti et al., 2016). Regarding splicing products, different digestion enzymes used in proteomics are biased toward identifying certain splicing junctions with small overlap among them (Wang et al., 2018). On the other hand, since transcript isoforms share a considerable proportion of their sequence, isoform-specific peptides are hard to identify (Tran et al., 2017). Nonetheless, proteogenomic approaches have improved the detection of peptides that can be uniquely matched to protein isoforms (Agosto et al., 2019). An added complexity in MS analysis is sequence identification from peptide mass spectra. Since the application of de novo peptide sequencing to MS remains challenging, the current analysis relies on database searches and can lead to false positives and neglect certain peptides (Bogdanow et al., 2016; Karunratanakul et al., 2019). In particular, a significant proportion of missed and unassigned peptides carry PTMs that shift the mass, which strongly impacts the database searches (Chick et al., 2015). Taken together, all these factors support the notion that there is an underestimation by MS experiments of the number of protein isoforms occurring in the cell.
3.2 Predicting the functional impact of alternative splicing from the sequenceIn parallel to the proteomics approaches, other studies have tried to address the functional impact of alternative splicing using computational models to predict the function of the resulting coding and protein sequences. The most straightforward approach has been to analyze the effect of splicing events on the coding sequence (CDS). For instance, in VastDB (Tapial et al., 2017), predictions are classified into ORF-preserving events, which have the potential to generate a fully functional protein, NMD targets, or ORF-disrupting events, including both frame-shifted and truncated CDS. However, these predictions are based on the impact of specific local events in isolation, without considering what happens elsewhere in the transcript, which does not reflect the complete splicing change. Indeed, a study of the functional impacts of isoform switches in cancer indicated that about 70% of these changes correspond to a single local splicing event. In contrast, the rest include changes that cannot be described in terms of the primary events (Climente-González et al., 2017). Addressing this issue, the method ISOTOPE calculates the modified ORFs resulting from all the splicing alterations in the gene to calculate possible novel epitopes in tumor samples (Trincado et al., 2021).
Other studies have measured the impact in terms of the alteration in functional domains. Such approaches map protein domains and functional elements to transcript isoforms to identify which functions are lost or gained due to AS (Hiller et al., 2005; Sulakhe et al., 2019; de la Fuente et al., 2020; Climente-González et al., 2017). For instance, one of these studies found that a large proportion of the isoform switches observed in cancer impacted functional domain families that are also targeted by cancer driver mutations (Climente-González et al., 2017). In a different study, integrating the functional annotations with differential transcript usage (DTU) showed that ~80% of the genes with DTU between two different mouse neural precursor cells present differences in the content of the functional features (de la Fuente et al., 2020).
The impact on protein structure can also be used to evaluate the potential functional variability resulting from AS. Variable exonic regions have been observed to generally code for disordered regions in proteins, which has been interpreted as enabling functional and regulatory diversity as disorder regions confer conformational flexibility that can be accommodated in the context of structured proteins (Hegyi et al., 2011; Romero et al., 2006). Modeling the impact of splicing on protein structure faces significant challenges since very few protein isoforms have a resolved 3D protein structure, and it remains difficult to obtain structures for proteins without a stable fold (Melamud & Moult, 2009). To address these challenges, Hao et al. (2015) implemented a semi-supervised learning approach based on structural information, and that does not require a negative set to conclude that 32% of alternatively protein isoforms result from SE events lead to functional proteins. Cross-species conservation has also been used as evidence for the functionality of protein isoform variants (Mazin et al., 2021; Rodriguez et al., 2013; Tapial et al., 2017). In fact, phylogenetically conserved isoform variants generally show higher expression from mRNA and a higher detection rate in proteomics experiments than nonconserved ones (Ezkurdia et al., 2012; Cummings et al., 2020).
3.3 Effects on functional networks of protein interactionsOne of the most intriguing functional effects of AS, and with potentially the farthest-reaching consequences, is the modulation of protein–protein interaction (PPI) networks. It has been proposed that proteins with associated tissue-specific splicing events are central hubs in interaction networks, having distinct interaction partners in different tissues (Buljan et al., 2012). Similarly, isoform switches in various cancer types have also been proposed to remodel PPIs involving cancer drivers and cancer-related pathways (Climente-González et al., 2017). Using in vitro experiments, it was observed that the inclusion of neuronally regulated exons significantly promoted or disrupted protein interactions using co-immunoprecipitation assays (Ellis et al., 2012). In addition, yeast-two-hybrid experiments revealed how to splice variants expand the network of interactions in the autism spectrum disorders (Corominas et al., 2014). This mechanism was further studied at genome-scale to reveal that less than 50% of interactions are shared among pairs of protein isoforms (Yang et al., 2016). Despite the vast range of the predicted isoform-specific interactions in tissues and disease conditions, most PPI databases only capture the interactome of canonical protein isoforms and describe these in terms of one protein per gene. Recently, the database DIGGER has provided a way to retrieve interactions between domains and proteins at the isoform and exon levels (Louadi et al., 2021). Understanding the rewiring of functional networks driven by AS can shed light on how even minor changes in the mRNA and protein sequences can trigger significant functional effects on the cell.
All the described studies have added multiple layers of evidence for an impact of the transcriptome variation on the proteome and its functional repertoire that appears to be more extensive than what can be directly observed from proteomics experiments alone. However, one could argue that even controlling for NMD or using additional structural and functional information, most studies have been based on computational predictions or in vitro experiments and require further experimental validation using cell or in vivo models, circling back to proteomics and its limitations. Below we describe alternative methodologies that address these limitations.
4 TRANSLATOMICS: FILLING THE GAP BETWEEN MRNA AND THE PROTEINThe basis for translatome analysis was already set more than 50 years ago (Warner et al., 1963; Steitz, 1969). However, it has been only recently that, thanks to combining those principles with high-throughput sequencing, translatomics has emerged as a field with the potential to capture the elements involved in mRNA translation in a transcriptome-wide manner (Zhao et al., 2019). We will focus on how translatomics techniques provide evidence of active translation of mRNAs and how this has been used to measure how AS can impact the proteome.
4.1 High-throughput measurement of translating mRNAsThe most widely used techniques for capturing translating mRNAs, polysome, and ribosome profiling, are based on the high-throughput sequencing of transcripts bound by ribosomes (Figure 3). Polysome profiling experiments are based on sucrose gradient separation and ultracentrifugation of full-length mRNAs, which have different sedimentation speeds according to the number of ribosomes sitting on them (Lackner et al., 2007; Mašek et al., 2011). This makes possible the stratification of different fractions containing mRNAs engaged by one (monosomes) or multiple (polysomes) ribosomes, indicating potentially different translation activities (Chassé et al., 2016; Floor & Doudna,
留言 (0)