
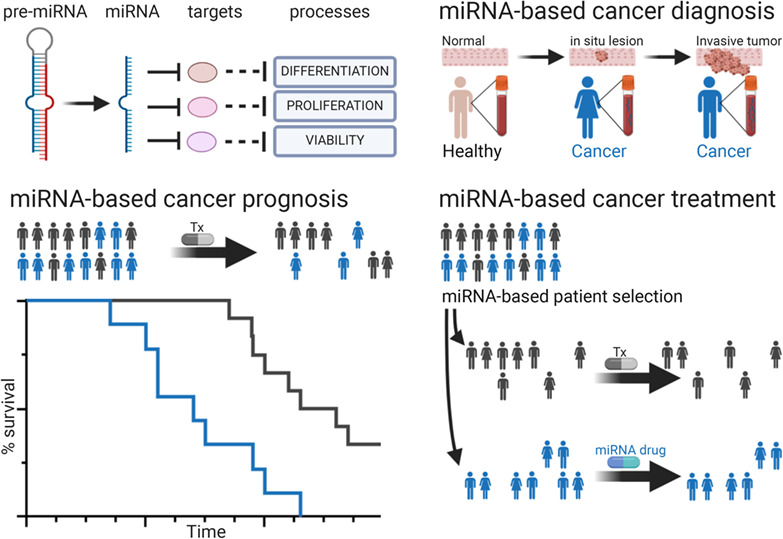
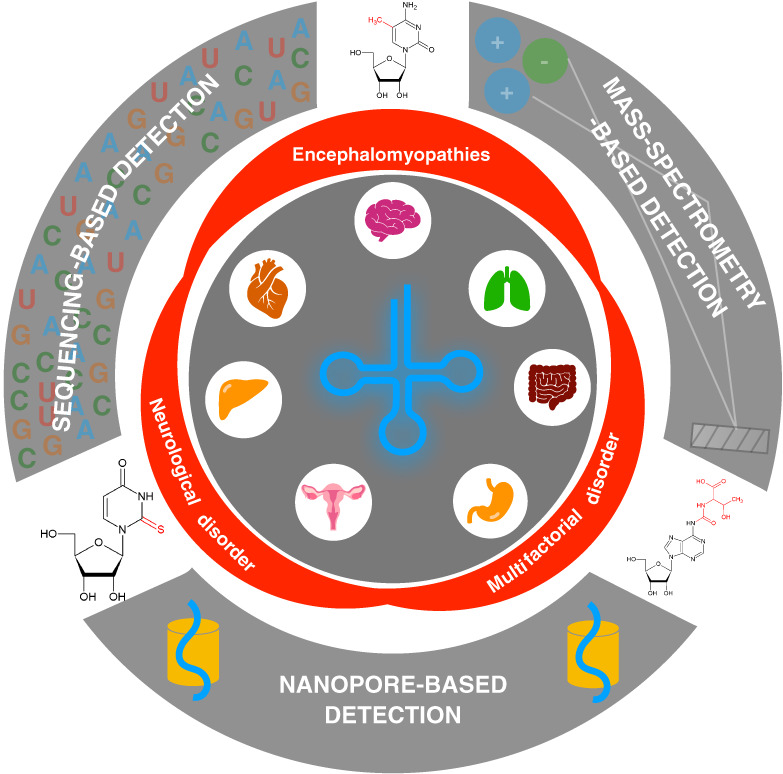
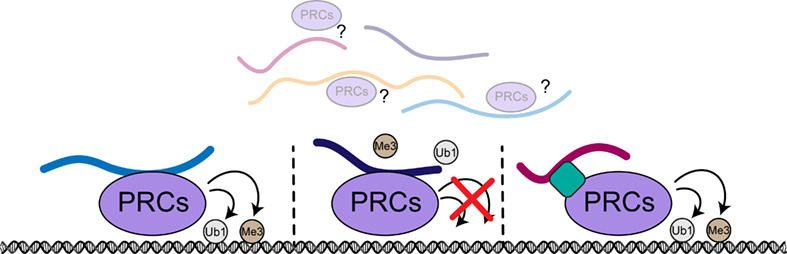
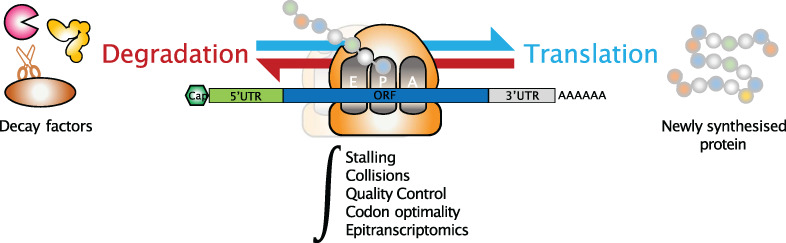
記住我
RNA modification in general, and RNA methylation in particular, have become a topic of intense research and publication activities during the past 10 years. This development has, in our opinion, been ignited by two seminal papers describing the distribution of N6-methyladenosine (m6A) residues in mammalian mRNA (Dominissini et al., 2012; K. D. Meyer et al., 2012), and has resulted in the reinvention of the RNA modification field as “epitranscriptomics” (K. D. Meyer & Jaffrey, 2014). While the modification field remains largely dominated by mRNA methylation, it has become clear that its biology cannot be disconnected from the remainder of modified RNA species (Ontiveros et al., 2020).
Preceding this development, our previous comprehensive review (Motorin & Helm, 2011) contained a minor section on mRNA, correspondingly larger sections on occurrence and function of RNA methylation in tRNA and rRNA, and outlooks on the topics of tRNA fragmentation and mitochondrial RNA methylation. Gratifyingly, the latter topics have grown into fields of their own right by now. While that review did indeed reflect the major developments in the preceding decade in a near-comprehensive fashion, we realize that we will be unable to repeat such an accomplishment without pointing to a number of reviews that cover important developments in the field. These include for example, modification detection techniques (Helm & Motorin, 2017; Limbach & Paulines, 2017; Linder & Jaffrey, 2019; Motorin & Marchand, 2021; Yoluc et al., 2021), complex modification machineries and their structure (Bourgeois et al., 2017; Lence et al., 2019), MTase target recognition, enzymatic mechanisms, impact of RNA methylation in numerous fields of biology and human pathology (Zaccara et al., 2019) including cancer research (Nombela et al., 2021), as well as immunology (Dalpke & Helm, 2012; Freund et al., 2019) and virology (Ruggieri et al., 2021). Structure and content of this update reflect our personal opinions on what the most important developments in the field were, and what we consider promising new angles.
A basic aspect here is the steadily climbing number of known RNA modification structures, within which methylations continue to be highly represented. Here we consider all modifications into which at least one C1 carbon unit has been incorporated either from the tetrahydrofolate cofactor, or from the more typical S-adenosyl-l-methionine (SAM). Most such incorporations occur directly on the nucleobase, or feature one or more linker atoms between nucleobase and C1 body. Examples for such structures are shown in (Figure 1a), and Figure 1b depicts methylations on the ribose. A particular situation is present for example, in wybutosine derivatives because the first step of its biosynthesis is a G37 methylation to m1G, although after the subsequent multiple enzymatic conversions, that carbon is barely recognizable as a C1 unit any more (Figure 1c). Of note, the other (non-C1) side chains of the sulfur atom in SAM are also metabolized, such as the 3-amino-3-carboxypropyl chain in acp-type modification of uridines (B. Meyer et al., 2020) or elsewhere in wybutosine, but these are not topic of this review. An impression of the multitude of methylated positions in the various nucleotides can be obtained from Figure 2.

Direct and indirect methylation of nucleobases (a), of the ribose (b) and the m1G37 methylation as an intermediate in the wybutosine (yW) pathway (c)

Diversity of nucleotide methylation. (a) Methylation sites on the chemical structures of the four major ribonucleotides, inosine, and pseudouridine. Methylation positions at carbon atoms are in green, at the cyclic and the exocyclic nitrogens are in light and dark blue, respectively. Position of 2′-O-methylation of the ribose residues are in yellow. Multiple modifications may occur sequentially on a single nucleotide. Secondary methylations of other modified residues in RNA, as well various derivatives of Y-base (yW) formed by -adenosyl-l-methonine (SAM)-dependent methyltransferases (MTases) are also included (shown here in italics). Reports on the existence of m22Gm, m62Am, m3Um, and m5D remain to be confirmed (shown in gray)
Also, one of the major aspects of epitranscriptome research was, and continues to be, the discovery and characterization of modification enzymes and their targets. An overview of the phylogenetic distribution in abundant RNA targets, that is, tRNA and rRNA is given in Figure 3.

Wien diagram for phylogenetic distribution of methylated nucleotides (found in tRNA (a) and rRNA (b) species) in major life domains. Secondary methylations and derivatives of Y-base (yW) are included
While sites in other targets, especially in those of low abundance, continue to be the subject of controversial discussion, advance in the identification of the so called epitranscriptomic “writers” is steady and reproducible with few exceptions (X. Zhang et al., 2012). Consequently, we report on numerous newly identified modification enzymes acting on tRNA and rRNA. This development focusses on higher eukaryotes and has enormously benefitted from early characterization of homologs in single cell model organisms (Bjork et al., 1987; Jackman et al., 2007; Maden & Hughes, 1997; Suzuki et al., 2007). We direct attention to a number of new protein complexes that were identified in eukaryotes. Three classes of particularly sophisticated MTase complex architectures are discussed in Box .
The variable composition of the Trm112-centered complexes supports the relatively new notion, that tRNA and rRNA methylation may constitute a physiological response to a stimulus, rather than being an obligate maturation step that inevitably leads to full stoichiometric methylation of a given site. This notion of dynamics is a recent development in the field of RNA methylation, and it comes in different flavors. In addition to dynamic reassembly of “writer” complexes, another flavor includes modification removal by “erasers” (Ontiveros et al., 2020). Some noteworthy facts on oxidative demethylation by “erasers” are given in Box , and more details on mRNA demethylation are laid out in the corresponding section on coding RNA.
Of note, although oxidative removal of methylgroups by erasers is an attractive concept in gene regulation (Zaccara et al., 2019), dynamic changes of methylation stoichiometry could plausibly originate from a combination of degradation and renewed synthesis, a factor that remains remarkably under-investigated. Occurrences and implications of partially methylated sites will be discussed in the subsequent sections, which are dedicated to tRNA, rRNA, and coding RNA. New developments in snoRNAs and other species are competently covered elsewhere (Hofler & Carlomagno, 2020).
2 TRANSFER RNA METHYLATIONTransfer RNA (tRNA) molecules display the greatest variety of RNA modifications in general, and in consequence, also of RNA methylations. Some of those methylations are highly conserved in different kingdoms (m5U54, Gm18, m1G37, and m7G46), while others are specific for a given branch or even group of species. Substantial efforts during the last 10 years allowed discovery of many new methylated sites in tRNA, in certain cases approaching comprehensive characterization of tRNA methylation profiles in different model organisms (Figure 4).

Methylated nucleotides in tRNA and their respective enzymes. Sites of nucleotide methylation on the tRNA cloverleaf structure are indicated for selected model organisms for bacteria (Escherichia coli), archaea (Halobacterium volcanii), and lower eukaryota (cytoplasmic tRNAs from Saccharomyces cerevisiae). For higher eukaryotes, a compilation of animal cytoplasmic tRNAs is shown. Hypermodifications that include methylation steps are also included, such as for example, various derivatives of the Y-base (yW). Universally conserved methylations are gray shaded, conserved in archaea and eukaryotes are in orange, and between bacteria and eukaryotes are in blue. m1ψ in archaeal tRNAs is considered similar to m5U
tRNAs from gram-negative bacterial species (e.g., Escherichia coli) may show methylations at seven different positions, and the same position within different tRNAs may bear different methylation types (e.g., at positions 32, 34, or 37). In comparison, Gram-positive bacteria lost Gm18 in the D-loop, but gained m1A22 in the same domain. Interestingly, the same highly conserved m5U54 is formed by two distinct enzymes in Gram-positive and Gram-negative bacteria. Gram-positive bacteria present the unique case of the tRNA methyltransferase TrmFO, which is folate-dependent instead of using the otherwise universal SAM cofactor (vide infra). The thermophilic bacterium Thermus thermophilus exhibits some extra tRNA methylations otherwise found in eukaryotes, namely m2G6 and m1A58, and the level of some tRNA methylations is regulated as a function of growth temperature (Kumagai et al., 1980; Ny & Bjork, 1980; Noon et al., 2003). Archaeal species show great diversity of tRNA methylation sites and many hypermethylated nucleotides (both base and 2′-O-methylations of the same nucleotide) are exclusively found in this living domain. Identification of the corresponding enzymatic activities is still ongoing in many instances, but essentially complete for some extensively studied species, like E. coli and Saccharomyces cerevisiae. Current knowledge on the tRNA methylations in different bacterial and archaeal species is summarized in Table 1.
TABLE 1. Bacterial and archaeal tRNA methylations E. coli (de Crecy-Lagard et al., 2020) B. subtilis (de Crecy-Lagard et al., 2020) T. thermophilus (Hori, 2019) Archaeaa tRNA position Modification Enzyme Modification Enzyme Modification Enzyme Modification Enzyme 6 m2G TrmN (TTC1157) Cm 9 m2G 9 m1G aTrm10 10 m2G/m2,2G aTrm-m22G (Trm11) 18 Gm TrmH Gm TrmH 22 m1A TrmK 26 m2G/m2,2G Trm1 27 m2G/m2,2G Trm1 32 Um/Cm TrmJ Cm 32 m5Cm 34 Um/Cm/U*m TrmL Gm/Cm CspR Cm Fib/sRNA 37 m1G TrmD m1G TrmD m1G TrmD 37 m6A TrmN6 (TrmG) m6A TrmN6 37 m2A RlmN m2A RlmN 39 Um Fib/sRNA 39 m5C 40 m5C 46 m7G TrmB/yggH m7G TrmB m7G TrmB 48 m5C aTrm4 49 m5C aTrm4 49 m7G 51 m5C 54 m5U(rT) TrmA m5U(rT) TrmFO m5U(rT) TrmFO m5U(rT) RlmD 54 m1ψ RlmY 56 Cm aTrm56 and Fib/snoRNA 57 m1A aTrmI 57 m1I aTrmI 57 m2G 58 m1A TrmI 67 m2G aTrm14 72 m5C aNSUN6Eukaryotic tRNA modification profiles are relatively well conserved from yeast to human (known eukaryotic activities listed in Table 2), with only a few extra sites detected in higher eukaryotic tRNAs (m2G5/6, m5C38, Gm/Ψm39, and m5C50/72). In contrast, eukaryotic mitochondrial tRNAs are only sparsely modified, with only few methylations found at positions 26, 37, and 54 (S. cerevisiae). Mammalian mitochondrial tRNAs contain m1A/m1G9, m2G10, m3C32, transiently formed m5C34, m5C48/49/50, and m1A58 in addition (Suzuki et al., 2020), (Suzuki & Suzuki, 2014).
TABLE 2. tRNA methylation in Saccharomyces cerevisiae and Homo sapiens S. cerevisiae H. sapiens tRNA position Cytoplasmic modification Cytoplasmic enzyme Mitochondrial modification Mitochondrial enzyme Cytoplasmic modification Cytoplasmic enzyme Mitochondrial modification Mitochondrial enzyme 4 Cm/Am Trm13 5 m2G 6 m2G 9 m2G 9 m1G Trm10 m1G TRMT10A m1G/m1A TRMT10C (MRPP1) 9 m1A TRMT10B 10 m2G Trm11 m2G TRMT11 m2G 14 m1Aa 18 Gm Trm3 Not present Gm TARBP1 20 m3Cb 26 m2G/m2,2G Trm1 m2G/m2,2G Trm1 m2G/m2,2G hTRM1c 27 m2,2G hTRM1 32 Cm Trm7/Trm732 Not present Um/Cm/ψm FTSJ1 or Fib/snoRNA 32 m3C Trm140 Not present m3C METTL2 and/or METTL6 m3C 34 Cm/U*m Trm7/Trm734 Not present Cm/f5Cm/Um/Gm FTSJ1 34 m5C Trm4 (Ncl1) m5C → hm5C → f5C NSUN3 37 m1G Trm5 m1G Trm5 m1G TRM5(TRMT5) 37 m1I Trm5 m1I TRM5(TRMT5) 38 m5C DNMT2 (TRDMT1) 39 Gm/ψm 40 m5C Trm4 (Ncl1) 44 Um Trm44 Um TRM44 (METTL19) 46 m7G Trm8/82 Not present m7G METTL1 (WDR4) 47d m3Cb 48 m5C Trm4 (Ncl1) Not present
留言 (0)