
記住我


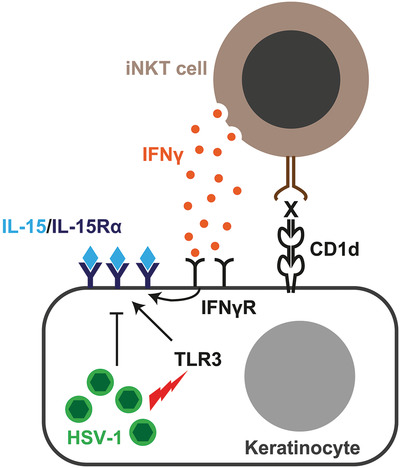






Herpes simplex virus type 1 (HSV-1) is a common pathogen which infects and persists in a majority of the adult human population worldwide [1]. HSV-1 is most commonly associated with cold sores, and can also cause severe, potentially fatal disease in neonates, the elderly, and immunocompromized patients [2, 3]. The immune response to HSV-1 infection is initiated by PRRs, including cytosolic DNA sensors, as well as TLRs 2, 3, and 9 [4]. Defects in TLR3 are associated with more severe HSV-1 infection, worse clinical prognosis, and development of encephalitis [5-7]. Despite vigorous innate and adaptive immune responses, HSV-1 can establish infection and persist in the human host. To accomplish this, the virus has evolved multiple mechanisms to avoid innate immunity [8], and evade MHC class I-mediated antigen presentation to CD8 T cells [9-12].
Interleukin 15 (IL-15) is expressed in many cell types and promotes proinflammatory responses against intracellular pathogens [13]. IL-15 functions in complex with the IL-15 receptor subunit α (IL-15R-α), to which it binds already in the ER [14]. In this form, it can be released as a soluble complex [15] or, exert its effects by cell surface transpresentation to immune cells expressing the IL-2/IL-15Rβ-γc [16]. The latter mechanism accounts for most of the effects of IL-15 on immune responses [17]. In the context of barrier immunology, IL-15 is critical for a variety of local tissue immune functions [18], and acts to activate multiple immune cell types, including NK cells [19, 20], CD8+ T cells [21], γδ T cells [22], and mucosa-associated invariant T cells [23]. IL-15 is expressed by many cell types, and in the context of skin, HSV-1-infected DCs stimulate CD8+ T-cell priming through IL-15 production [24].
Invariant natural killer T (iNKT) cells are a distinct type of innate-like T cells, characterized by an invariant TCR-α chain Vα24-Jα18 rearrangement and restriction by the MHC class I-related molecule CD1d. These cells recognize glycolipid antigens presented by CD1d and upon activation rapidly release cytokines characteristic of both Th1 and Th2 responses [25, 26]. iNKT cells play an important role in the immune response to HSV-1, as mice lacking either CD1d or iNKT cells show an impaired immune response against the virus [27, 28]. The importance of iNKT cells in the immune response to HSV-1 infection is further underscored by the finding that HSV-1 inhibits CD1d-mediated antigen presentation in DCs, thus, interfering with the iNKT cell response [29-32]. Furthermore, HSV-1 also inhibits iNKT cell activation by a CD1d-independent mechanism, dependent on direct cell-to-cell contact [33].
In this study, we found that expression of the IL-15/IL-15R-α complex in response to HSV-1 infection is characterized by dynamic kinetics, with initial upregulation by TLR3-mediated recognition of virus, followed by active downregulation by the virus. This gives the immune system a short time window to respond to the IL-15 activation signal. However, the kinetics can be modulated by iNKT cells through the release of IFN-γ. The action by iNKT cells slows down and counteracts the viral downregulation of the IL-15/IL-15R-α complex. Thus, we identify a novel HSV-1 immune evasion strategy and find that iNKT cells can inhibit this mode of viral immune evasion.
Results HSV-1 infection triggers surface upregulation of IL-15 and IL-15R-α in keratinocytesHSV-1 infection is known to induce upregulation of IL-15 in human monocytes [34]. To test whether this occurs also in keratinocytes, the first cell type infected in the course of primary HSV-1 infection, we infected HaCaT cells, which have the capacity to produce IL-15 [35]. Surface expression of both IL-15 and IL-15R-α was upregulated in response to infection (Fig. 1A). Some induction of IL-15 occurred already at 6 h after infection, and both IL-15 and IL-15R-α increased over time, reaching four to six times the baseline levels at 18 h after infection (Fig. 1B). To investigate if this upregulation was linked with induction of gene expression, we harvested RNA from HaCaT cells infected with HSV-1 for 1–3 h and measured IL15 and IL15RA transcript levels by qPCR (Fig. 1C). There was an increase in both IL15 and IL15RA mRNA levels at 3 h after infection. Together, these results indicate that the increase in IL-15 and IL-15R-α expression in HSV-1-infected keratinocytes occurs at both surface protein and mRNA levels.

The IL-15/IL-15R-α complex expression is induced upon HSV-1 infection, at both protein and mRNA levels. (A) Representative histograms of IL-15 and IL-15R-α surface expression in uninfected and HSV-1-infected HaCaT keratinocytes at 18 h after infection. (B) IL-15 and IL-15R-α surface protein expression kinetics during the first 18 h after HSV-1 infection (MFI relative to uninfected control; mean ± SD from six biological replicates assessed in three independent experiments). HSV-1-infected HaCaT cells in cultures were defined by flow cytometry surface staining for the HSV glycoprotein D (gD). (C) IL15 and IL15RA gene expression induction in the early hours after HSV-1 infection, relative to uninfected (mean ± SD from 12 biological replicates assessed in four independent experiments). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s., not significant. Statistics calculated using Mann-Whitney U test.
Induction of the IL-15/IL-15R-α complex is dependent on TLR3As TLRs are known to be involved in the recognition of HSV-1, we next aimed to determine the role of such receptors in the induction of IL-15R-α expression in HSV-1-infected keratinocytes. To investigate this, HaCaT cells were preincubated with 1 μM Bafilomycin A1, an inhibitor of autophagy and, thus, an inhibitor of the function of TLRs 3, 4, 7, and 9, for 1 h prior to infection with HSV-1 for 15 h (Fig. 2A). Bafilomycin A1 partially inhibited the induction of IL-15R-α expression in response to HSV-1 infection, suggesting TLR involvement. To better define which TLR was involved in IL-15R-α upregulation, the expression of TLRs 1–10 was assessed in HaCaT cells using qPCR (Fig. 2B, Supporting Information Fig. S1). The HaCaT cells had detectable expression of TLRs 2, 3, 5, 6, and 10, whereas transcripts for TLRs 1, 4, 7, 8, and 9 were not detected. Given that it is known that HSV-1 can be detected by TLRs 2, 3, and 9 [4], we next evaluated if ligands for these receptors could trigger the induction of IL-15R-α. To test this, HaCaT cells were incubated for 15 h with the TLR2 ligand zymosan, the TLR3 ligand poly(I:C), or the TLR9 ligand dsDNA-EC, each at 1 μg/mL (Fig. 2C). Among these three TLR ligands, only the TLR3 ligand poly(I:C) induced an enhanced level of surface IL-15R-α expression.

Upregulation of IL-15 and IL-15R-α is mediated by TLR3. (A) Inhibition of IL-15R-α upregulation by bafilomycin A1 (BafA1) (MFI relative to uninfected control; mean ± SD from six biological replicates assessed in three independent experiments). (B) TLR expression in HaCaT cells, normalized to GAPDH. (C) Surface expression of IL-15R-α after 18 h stimulation with TLR ligands (MFI relative to untreated control; mean ± SD from six biological replicates assessed in three independent experiments). dsDNA-EC: Double-stranded genomic DNA from E. coli K12 (D) Inhibition of IL-15R-α surface protein upregulation by CU CPT 4a and TPCA-1 at 15 h after infection (MFI relative to uninfected control; mean ± SD from three biological replicates assessed in two independent experiments). (E) Inhibition of IL15RA mRNA upregulation by CU CPT 4a and TPCA-1 at 3 h post HSV-1 infection or poly(I:C) treatment (relative to untreated control; mean ± SD from seven biological replicates assessed in four independent experiments). *p < 0.05, **p < 0.01, ***p < 0.001. n.s., not significant. Statistics calculated using Mann-Whitney U test.
To ascertain if TLR3 was involved in HSV-1 recognition, HaCaT cells were treated with the TLR3 inhibitor Cu CPT 4a or with IκB kinase inhibitor TPCA-1 (both at 1 mM) for 1 h prior to HSV-1 infection. Cells were harvested after 15 h for analysis by flow cytometry. Both Cu CPT 4a and TPCA-1 potently inhibited the HSV-1-induced IL-15R-α upregulation (Fig. 2D). To investigate whether this inhibition could be seen at the transcriptional level, we next measured mRNA levels at an early time point postinfection. HaCaT cells were treated with either Cu CPT 4a or TPCA-1 for 1 h prior to HSV-1 infection or poly(I:C) treatment, and cellular RNA was harvested after 3 h (Fig. 2E). Cu CPT 4a and TPCA-1 both strongly inhibited HSV-1-induced IL15RA mRNA expression, and at least partially inhibited poly(I:C)-induced IL15RA mRNA expression. Together, these findings indicate that HSV-1-induced upregulation of IL-15R-α is dependent on TLR3.
HSV-1 interference with surface expression of IL-15 and IL-15R-αTo determine whether the elevated levels of IL-15 and IL-15R-α are maintained in HSV-1-infected cells over time, surface levels were measured by flow cytometry throughout a 36-h infection time course (Fig. 3A). Surface levels of both IL-15 and IL-15R-α peaked after 18 h of infection, after which expression of both proteins started to drop, reaching levels well below baseline after 24 h and not returning to baseline throughout the entire 36 h period. To investigate if this effect was also evident on the transcriptional level, we harvested RNA from infected cells at consecutive time points throughout the 36-h infection time course and measured IL15 and IL15RA mRNA levels. Transcript levels of both IL15 and IL15RA followed a similar pattern as surface protein expression, with mRNA levels peaking at 3 h after infection dropping below baseline levels after 12 h (Fig. 3B). In uninfected HaCaT cells, the IL15 and IL15RA transcripts remained expressed at similar levels throughout the time course (Supporting Information Fig. S2). The GAPDH transcript was detectable throughout as well, in both HSV-1-infected and -uninfected cells (Supporting Information Fig. S2).

IL-15 and IL-15R-α are downregulated by a viral replication-dependent mechanism. (A) Time course of surface IL-15 and IL-15R-α expression in HSV-1 infection 0–36 h (MFI relative to uninfected; mean ± SD from nine biological replicates assessed in six independent experiments). (B) Time course of IL15 and IL15RA mRNA expression in HSV-1 infection 0–36 h (relative to uninfected; mean ± SD from seven biological replicates assessed in three independent experiments). (C) Degradation of intracellular IL-15 and IL-15R-α in HSV-1 infection after 48 h (MFI relative to uninfected; mean ± SD from five biological replicates assessed in three independent experiments). (D) Lack of poly(I:C)-induced downregulation of surface IL-15R-α (MFI relative to uninfected; mean ± SD from three biological replicates assessed in two independent experiments). (E) Inhibition of IL-15 and IL-15R-α downregulation by PAA at 42 h after infection (MFI relative to uninfected; mean ± SD from three biological replicates assessed in two independent experiments). (F) Inhibition of IL-15 and IL-15R-α downregulation by acyclovir at 42 h after infection (MFI relative to uninfected; n = 3). In (A) and (C) identification of HSV-1-infected HaCaT cells was performed by surface staining for the HSV glycoprotein D (gD). *p < 0.05, **p < 0.01, ***p < 0.001. n.s., not significant. Statistics calculated using Mann-Whitney U test.
To determine if the IL-15R-α/IL-15 complex at later infection time points may be recycled into the cell or actively degraded, we infected HaCaT cells for 48 h and measured intracellular IL-15 and IL-15R-α levels by intracellular flow cytometry (Fig. 3C). While surface IL-15 and IL-15R-α levels in HSV-1-infected cells were modestly downregulated and remained at around 70% of baseline, intracellular IL-15 content was more severely depleted to levels below 10% of baseline, and IL-15R-α to levels around 40% of baseline, respectively, indicating that loss from the cell surface did not merely reflect internalization and that intracellular protein degradation may be involved. Since the IL-15/IL-15R-α complex can also be released into the medium, we measured IL-15 levels in the supernatant over a 36-h infection time course by ELISA (Supporting Information Fig. S3). Despite high variability throughout the time course, we found that IL-15 levels in the supernatant of HSV-1-infected cells were approximately 10-fold lower than those seen with control cells, suggesting that shedding was not a major contributor to the loss from the cell surface.
To investigate whether the loss of IL-15 and IL-15R-α was part of normal cellular dynamics following the initial upregulation, or if the downregulation was specific to HSV-1 infection, we treated HaCaT cells with poly(I:C) for 18 and 36 h (Fig. 3D). When stimulated in this way, IL-15R-α was first upregulated and then gradually returning, but not dropping below baseline values. We next investigated whether the loss of IL-15 and IL-15R-α expression in HSV-1-infected cells was dependent on active viral replication. To that end, we treated HaCaT cells with the viral DNA polymerase inhibitors phosphonoacetic acid (PAA) (at 4 mM) or acyclovir (at 1 mM) for 1 h before HSV-1 infection for 42 h (Fig. 3E and F). Both PAA and acyclovir inhibited the loss of surface IL-15 and IL-15R-α in infected cells. Together, these results indicate that the expression of IL-15 and IL-15R-α in HSV-1-infected HaCaT cells is actively downregulated by a mechanism dependent on viral replication.
iNKT cells counteract downregulation of IL-15 and IL-15R-α in an IFNγ-dependent mannerHSV-1 infection is known to downregulate CD1d expression in DCs, thus, counteracting iNKT cell activation [29-33]. In contrast, we previously found that CD1d-down regulation does not occur in primary human keratinocytes infected with HSV-1 [33]. To investigate if CD1d downregulation occurs in HaCaT cells, HaCaT cells were infected with HSV-1 for 6, 18, or 42 h and surface CD1d levels were measured using flow cytometry (Fig. 4A and B). At all the measured time points, CD1d was not significantly downregulated in infected HaCaT cells.

Invariant NKT cells counteract HSV-1 induced downregulation of IL-15/IL-15R-α via IFN-γ release. (A) Flow cytometric measurement of CD1d surface expression in HaCaT cells after 6, 18, or 42 h of HSV-1 infection. (B) CD1d surface expression in HaCaT cells after 6, 18, or 42 h of HSV-1 infection (MFI relative to uninfected; mean ± SD from two biological replicates assessed in two independent experiments). (C) Concentration of IL-4 and IFN-γ in the supernatant of iNKT cells cocultured with HaCaT cells infected with HSV-1 or pulsed with 1 ng/mL αGalCer for 18 h (mean ± SD from nine biological replicates representing three independent experiments; statistical significance against uninfected control). (D) IL-15R-α surface expression in HaCaT cells after a 42 h HSV-1 infection, with or without iNKT cells present (MFI relative to 18 h postinfection; mean ± SD from six biological replicates assessed in four independent experiments). (E) Progress of HSV-1 infection determined by flow cytometry staining for HSV glycoprotein D (gD) in HaCaT cells with or without iNKT cells present (mean ± SD from six biological replicates assessed in two independent experiments). (F) Percentage of dead cells among HSV gD positive and negative HaCaT cells at 18 and 42 h postinfection with or without iNKT cells (mean ± SD from 6 biological replicates representing 3 independent experiments). *p<0.05, **p<0.01. n.s., not significant. Statistics calculated using Mann-Whitney U test.
It is known that expression levels of ligands of NKG2D can be modulated by HSV-1 infection [36, 37]. This is relevant in the context of iNKT cells, since NKG2D can contribute to iNKT cell activation [38]. We, therefore, measured levels of NKG2D ligands MICA, MICB, ULBP2, and ULBP3 on cell surface of infected HaCaT cells after 18 and 42 h after infection (Supporting Information Fig. S4). In addition, we measured receptors important for the regulation of the immune response including HLA-ABC, CD155, CD112, EpCAM, CD54, CD102, and CD50. Most of these receptors were upregulated, in line with an ability for infected HaCaT cells to recruit and activate the cellular immune response including iNKT cells. Interestingly, one of the NKG2D ligands tested, ULBP3, showed a similar pattern of surface up- and downregulation as the IL-15/IL-15R-α complex. In contrast, ULBP2 showed signs of downregulation, while MICA expression was elevated at the late time point.
Previous findings have suggested a role for iNKT cells in immune control HSV-1 infection [27], and we hypothesized that iNKT cells may be able to counteract the virus-induced IL-15/IL-15R-α complex downregulation. To evaluate this possibility, iNKT cells were cocultured with HaCaT cells infected with HSV-1 or pulsed with α-galactosylceramide, and IFN-γ and IL-4 was assessed in the supernatant. HSV-1-infected HaCaT triggered production of IFN-γ while IL-4 could not be detected (Fig. 4C). This was in contrast to α-galactosylceramide, which triggered production of both cytokines. Next, HaCaT cells were infected with HSV-1 and cultured alone or in the presence of iNKT cells (Fig. 4D, Supporting Information Fig. S5). Interestingly, the presence of iNKT cells in the culture supported continuous IL-15R-α HaCaT surface expression levels at around three times the preinfection baseline levels even after 42 h of infection. To evaluate if IFN-γ produced by the iNKT cells may mediate this effect, blocking with anti-IFNγ was performed. The effect of iNKT cells was inhibited by the IFN-γ antibody, indicating a role for this cytokine in the maintenance of elevated IL-15/IL-15R-α complex surface levels in HSV-1- infected HaCaT cells.
We were next interested to evaluate if iNKT cells could inhibit HSV-1 infection in keratinocytes exposed to the virus. HaCaT cells were infected with HSV-1 for up to 42 h in the presence or absence of iNKT cells, and the percentage of cells positive for HSV-1 envelope gp D (gD) on the cell surface was assessed as a measure of active infection (Fig. 4E). Throughout the infection timeline, the presence of iNKT cells reduced the number of HaCaT cells positive for gD, suggesting an antiviral effect. However, there was no indication of increased cell death among HaCaT cells in the coculture in the presence of iNKT cells, supporting a role for a nonlytic effect by iNKT cells (Fig. 4F). Altogether, these data indicate that iNKT cells play a role in the innate immune response to HSV-1 at the keratinocyte level, counteracting virus-induced downregulation of the IL-15/IL-15R-α complex via an IFNγ-dependent mechanism, as well as slowing down the viral infection progress.
DiscussionIL-15 acts to activate and recruit innate and adaptive immune cells to a local site of infection. In this study, we found that cell surface expression of the IL-15/IL-15R-α complex is induced and rapidly upregulated in keratinocytes in response to HSV-1 infection. This IL-15 expression is characterized by dynamic kinetics, with initial upregulation in response to TLR3-mediated recognition of HSV-1, followed by active downregulation by a viral immune evasion mechanism. This may give the immune system a rather brief time window to recognize and respond to the IL-15 activation signal induced by the virus. Moreover, the rapid kinetics are modulated by iNKT cells through the release of IFN-γ, which slows and inhibits the viral downregulation effect such that the IL-15/IL-15R-α complex is maintained on the cell surface. The iNKT cells also act to inhibit HSV-1 replication in infected cells. These findings identify a novel HSV-1 immune evasion strategy and illustrate how iNKT cells can inhibit this mode of viral immune evasion (Fig. 5).

Summary of IL-15 dynamics in response to HSV-1 infection in human keratinocytes and the effect of iNKT cells in this system. The figure illustrates the keratinocyte cellular response to HSV-1 infection with TLR3-dependent upregulation of cell surface IL-15R-α/IL-15 complexes, which is followed by rapid viral inhibition of IL-15R-α/IL-15 expression and downregulation from the cell surface. This mode of viral immune evasion can in turn be counteracted by IFN-γ produced by iNKT cells. The X denotes a hypothetical endogenous CD1d-presented ligand.
IL-15 plays important roles in both innate and adaptive immunity in tissues [18]. In skin, hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T-cell homeostasis, and follicle-derived IL-15 is required for the establishment of resident memory CD8 T-cell responses in skin [39]. In the context of HSV-1 infection, IL-15 is important for both innate NK cell responses [40] and the formation of effective CD8 T-cell responses to the virus [24]. It, therefore, makes immunological sense for HSV-1 to target the IL-15/IL-15R-α complex for downregulation, and potentially for intracellular degradation, to evade recognition by NK cells and CD8 T cells recruited to the vicinity of the infected cell. Given that IL-15 acts mostly via cell surface transpresentation, its downregulation from the surface of actively infected cells may allow production of viral progeny even in the context of an ongoing immune response in the tissue.
Many viruses target the CD1d antigen presentation pathway, suggesting that iNKT cells play important roles in immune defense against such viral infections (reviewed in [41]). Most viruses probably do not encode CD1d-presented antigens, so the rationale for targeting CD1d is not self-evident. However, iNKT cells can be activated by endogenous CD1d-presented ligands upregulated in response to TLR-mediated cellular sensing of bacterial and viral infections [42, 43]. This mode of recognition has to our knowledge not yet been shown for HSV-1 and, thus, remains hypothetical, although plausible, in this case. For HSV-1 infection, our data indicate that downregulation of CD1d is cell-type dependent, as downregulation occurs in DCs but not in keratinocytes. This observation is consistent with the possibility that iNKT cells may play a bigger role in immune sensing of HSV-1 in keratinocytes. In support of this notion, we observe that iNKT cells can inhibit the HSV-mediated downregulation of the IL-15/IL-15R-α complex, and that this effect is at least partly mediated by IFN-γ. Interestingly, this effect occurs despite the observation that infected keratinocytes partly inhibit iNKT cell activation in a contact-dependent manner [33]. The iNKT cells, thus, act as an innate immunity sensor and countermeasure, which may prolong the window of opportunity for IL-15-dependent activation and recruitment of innate and adaptive effector cells to the site of infection. In addition, it is interesting to note that IFN-γ was previously shown to enhance TLR3 expression [44], and to suppress HSV-1 replication in keratinocytes [45], consistent with the role of iNKT cell-produced IFN-γ we observe here.
While the present study identifies a novel mode of HSV-1 immune evasion and a role for iNKT cells in countering this, the study also has several limitations. First, we do not yet know the viral factor responsible for downregulation of the IL-15/IL-15R-α complex. Finding the viral protein responsible will be important for detailed mechanistic studies and development of possible new antivirals targeting this mechanism. Second, we do not know if cognate CD1d-presented antigen recognition is needed for the effect of iNKT cells. Third, while we find that IFN-γ is involved in mediating the effect of iNKT cells, we do not know exactly how and if other iNKT cell functions are important. Future studies should address these open questions.
In summary, the findings in this study extend our understanding of the immune response against HSV-1 and the repertoire of immune evasion mechanisms employed by this virus. Furthermore, the findings indicate a role for iNKT cells in counteracting the immunoevasive downregulation of the IL-15 pathway. Given that TLR3-mediated sensing of viruses, as well as the CD1d-iNKT cell axis, are evolutionarily conserved pathways, the findings may be relevant in several infectious disease settings.
Materials and methods Ethics statementWritten informed consent was obtained from all healthy blood donor participants in accordance with study protocols conforming to the provisions of the Declaration of Helsinki and approved by the Regional Ethics Review Board in Stockholm (approvals 2007/772-32 and 2016/1415-32).
Invariant NKT cell culturesPeripheral blood collected from healthy donors recruited at the Blood Transfusion Clinic at the Karolinska University Hospital Huddinge was sampled and screened for iNKT cell frequency (defined as the frequency of Vα24+Vβ11+ cells in CD3+ population) by flow cytometry. PBMCs were isolated from blood where iNKT cell frequency exceeded 0.1%, using Ficoll-Hypaque density gradient centrifugation (Axis Shield Diagnostics, Dundee, United Kingdom). PBMCs were resuspended at 2 × 106 cells/mL in RPMI medium supplemented with 10% FCS (Atlas Biologicals, Fort Collins, CO, USA), 1 mM HEPES, and l-glutamine (RPMI-10), as well as 10 ng/mL IL-2 (PeproTech, Rocky Hill, NJ, USA) and 100 ng/mL of αGalCer derivative 7DW8-5 (Funakoshi, Tokyo, Japan) to stimulate iNKT cell expansion. After 14 days of expansion, iNKT cells were isolated using anti-Vα24 PE antibody, followed by positive selection with MACS anti-PE microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany).
Enriched iNKT cells were maintained in coculture with monocytes. Monocytes were obtained from peripheral blood by negative selection with RosetteSep human monocyte enrichment cocktail (STEMCELL Technologies, Vancouver, Canada), followed by isolation using Ficoll-Paque density gradient centrifugation. Monocytes were then resuspended in RPMI-10 at 2 × 106 cells/mL and subjected to irradiation for a total dose of 35 Gy. Invariant NKT cells were mixed with irradiated monocytes at 1:1 iNKT:monocyte ratio, and then resuspended at 2 million cells/mL in RPMI-10 supplemented with 10 ng/mL IL-2 and 100 ng/mL 7DW8-5. The cells were restimulated with fresh irradiated monocytes every 14 days. Medium volume was adjusted throughout the culture period to keep cell density at 2 × 106 cells/mL. To avoid mycoplasma contamination, iNKT cell, HaCaT, and Vero cell cultures were regularly tested using Universal Mycoplasma Detection Kit (ATCC, Manassas, VA, USA) according to the manufacturer's instructions.
Infection assaysConfluent monolayers of HaCaT cells grown in U-bottom microplates were infected with DMEM containing HSV-1 at MOI of 1 PFU/cell for 1 h at 37°C. After infection, cells were washed with PBS and incubated in DMEM medium containing 10% FCS. This was considered at time point 0 and from then on, cells were kept in the medium for up to 48 h. At designated time points, cells destined for flow cytometric analysis were detached from the surface using PBS supplemented with 1 mM EDTA and 1% FCS at 37°C. Cells destined for qPCR analysis were lysed directly in the culture plate and RNA was extracted using RNeasy Mini Kit (QIAGEN, Hilden, Germany) according to manufacturer's instructions.
Coculture experimentsInvariant NKT cells were washed in PBS and cultured in RPMI medium with 10% FCS, but without 7DW8-5 or IL-2, for at least 48 h prior to the start of HaCaT and iNKT cell coculture, to minimize effects of iNKT cell activation in growth culture. Confluent monolayers of HaCaT cells grown in U-bottom microplates were infected with HSV-1 as described above. After 1 h of infection, the cells were washed with PBS and iNKT-containing medium was added for an iNKT:HaCaT ratio of 2:1. The plates were centrifuged at 900 rpm for 3 min to promote direct cell-to-cell contact, then cultured at 37°C for the appropriate time. For analysis of proteins in coculture supernatants, the plates were centrifuged at 900 rpm for 3 min for the cells to collect at the bottom of the well and supernatant was collected. Supernatant was kept at −80°C until analysis. For flow cytometry, CD3 expression and cell size were used to distinguish between iNKT cells and HaCaT cells.
Ligands and inhibitorsTLR ligands zymosan, poly(I:C) (polyinosine-polycytidylic acid), and dsDNA-EC (double-stranded genomic DNA from E. coli K12) were obtained from Invivogen (San Diego, CA, USA). For TLR stimulation, HaCaT cell layers were washed and DMEM medium with 10% FCS containing the appropriate ligand was added at time point 0. The specific TLR3 inhibitor CU CPT 4a (N-[(3-Chloro-6-fluorobenzo[b]thien-2-yl)carbonyl]-d-phenylalanine) and IκB kinase inhibitor TPCA-1 (2-[(Aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide) were obtained from Bio-Techne Ltd. (Abingdon, United Kingdom). Autophagy inhibitor Bafilomycin A1 was obtained from Invivogen. Viral DNA polymerase inhibitors PAA and acyclovir were obtained from Sigma-Aldrich (St. Louis, MO, USA) and Abcam (Cambridge, United Kingdom), respectively. IFN-γ blocking antibody was obtained from Invivogen. For infection and treatment assays with inhibitors, HaCaT cell layers were washed with PBS, and serum-free DMEM medium containing the appropriate inhibitor was added for 1 h prior to the start of infection or treatment. In case of later medium changes, inhibitors were added to the medium at each change using the starting concentration.
Antibodies and flow cytometryFlow cytometry mAbs used in the project are listed in Supporting Information Table S1. Cell surface staining was performed using directly conjugated antibodies and fixed using Cytofix/Cytoperm or Transcription Factor Fixation/Permeabilization buffer (both from BD Biosciences, Franklin Lakes, NJ, USA) as appropriate. Intracellular staining was performed using the relevant mAbs in Perm/Wash or Transcription Factor Perm/Wash buffer as appropriate (both from BD Biosciences). Identification of HSV-1-infected HaCaT cells was performed by surface staining for the HSV gD where appropriate (ViroStat, Westbrook, ME) (Supporting Information Fig. S5). Samples were acquired on an LSRFortessa 18-colour flow cytometer (BD Biosciences) equipped with 405, 488, 561, and 639 nm lasers. Single-stained polystyrene beads (BD Biosciences) were used for compensation purposes. Software-based compensation was performed using the compensation platform in FlowJo software version 10.2 (FlowJo LLC, Ashland, OR, USA). For IL-15 and IL-15R-α staining, control stains with appropriate isotype controls (R&D Systems) were performed in parallel and isotype control signal values were subtracted from respective antibody signal values, to control for any increase in nonspecific antibody binding in HSV-1-infected cells.
qPCRRNA extracted from the cells was treated with Ambion DNase I (ThermoFisher Scientific, Waltham, MA, USA) to remove any DNA contamination. Afterwards, 500 ng of RNA from each sample was synthesized to cDNA using Invitrogen SuperScript VILO cDNA Synthesis Kit (ThermoFisher Scientific), according to the manufacturer's instructions. qPCR analysis was done using the QuantiTect SYBR Green PCR kit (QIAGEN), according to the manufacturer's instructions, on a QuantStudio 5 Real-Time PCR system (ThermoFisher Scientific). GAPDH was used as endogenous control. Primer pairs used for amplification of IL15, IL15RA, and GAPDH were qSTAR qPCR Primer Pairs (OriGene Technologies, Rockville, MD, USA). Primer sequences are listed in Supporting Information Table S2.
Soluble protein analysisCollected and frozen cell culture supernatants were thawed on ice prior to analysis and stained using the ProcartaPlex Human Cytokine Panel 1B kit (ThermoFisher Scientific). The plate was read using a Bio-Plex MAGPIX Multiplex Reader machine (Bio-Rad, Hercules, CA, USA) and resultant readings were analyzed using Bio-Plex Manager MP software version 4.1 (Bio-Rad).
TLR analysisTo quantify the expression levels of various TLRs in HaCaT cells, cDNA samples obtained from uninfected cells as described above were analyzed by RT-PCR using the TLR RT-Primer Set (Invivogen), used together with Fermentas PCR Master Mix (ThermoFisher Scientific), with cycling parameters set according to the manufacturer's instructions, using the Veriti Thermal Cycler (ThermoFisher Scientific). Samples, thus, obtained were mixed with DNA loading buffer (Bio-Rad,), and run on a 3% Agarose (Sigma-Aldrich) gel containing GelGreen Nuclear Acid Gel Stain (Biotium, Fremont, CA, USA). The gel was then scanned using LAS-4000 Luminescent Image Analyzer (Fujifilm, Tokyo, Japan) and expression levels of each TLR estimated were based on the resulting image using ImageJ version 1.52a. GAPDH was used as endogenous control.
Virus stocksHSV-1 (strain F) stocks were prepared by infecting 80–90% confluent monolayers of African green monkey kidney Vero cells. Infected cells were incubated in DMEM (Invitrogen) supplemented with 5% FCS and harvested after2 days and subjected to two freeze-thaw cycles to lyse cells. After centrifugation at 20,000 × g for 10 min to pellet cell debris, clarified supernatant was aliquoted, and stored at −80°C until use. Virus titers were determined by plaque assay on Vero cell monolayers. Briefly, 10-fold dilutions of virus were adsorbed onto Vero cells for 1 h at 37°C. The inoculum was then removed and fresh medium containing 2% low melting point Agarose (Sigma) was added. Two days later, cells were fixed with 4% formalin and stained with 0.1% crystal violet solution to determine the number of plaques.
Statistical analysisDifferences between independent samples were assessed using Mann-Whitney U test, calculated based on raw data (prenormalization). Statistical analyses were performed using Prism version 7.0c for Mac OS X (GraphPad Software, La Jolla, CA, USA), and two-sided p-values < 0.05 were considered significant.
AcknowledgmentsThis research was supported by grants to JKS from the Swedish Research Council (2020-01743), the Swedish Cancer Society (20-1142PjF), and Center for Innovative Medicine (20190732).
Conflict of InterestThe authors declare no commercial or financial conflict of interest.
Author contributionsJ.K.S. and L.B. conceived the original research design.
M.J.S., D.P.P., M.M., M.S.C. and J.K.S. designed experiments.
M.J.S. conducted experiments.
M.J.S. analyzed experimental data.
M.J.S. and J.K.S. wrote the manuscript.
All authors reviewed and commented on the manuscript.
留言 (0)