
記住我








Severe corona disease 2019 (COVID-19) affects not only the lungs but many other organ systems including the central nervous system (CNS) [1, 2]. Similarly, long-term sequelae of COVID-19 also involve multiple peripheral tissues including the CNS [3]. While sampling from the respiratory microenvironment through bronchiolar lavage has facilitated our understanding of local processes in the lung during severe COVID-19 [4, 5], less is known with regard to other organ systems. Such studies often rely on analysis of autopsy tissue material [6] but have nevertheless recently yielded insights into possible local pathophysiological mechanisms including the CNS [7]. An alternative approach, assessment of circulating immune cells, and/or soluble factors often fails to adequately provide information on events occurring in local microenvironments. In this regard, large-scale in-depth metabolomics and proteomics approaches might be detailed enough to reveal specific imprints from affected tissues [6]. Indeed, since circulating metabolomics provides a functional readout of cellular processes [8, 9], it is not surprising that changes in serum metabolites associate with COVID-19 severity [10-12]. However, drawbacks with many of these studies involve not correcting for underlying confounding factors such as comorbidities, which indirectly might affect the metabolome of an individual, as well as administered drugs and other xenobiotics, which more directly affect deep and sensitive metabolomic measurements [13]. Additionally, it has been challenging for multi-omics approaches integrating metabolomics with other large data quantities to associate alterations in the circulation with events occurring in tissue microenvironments. To address these outstanding questions, we here performed a deep metabolomic assessment in hospitalized moderate and severe COVID-19 patients and integrated the results with large-scale proteomic and flow cytometry data to capture multisystem perturbations.
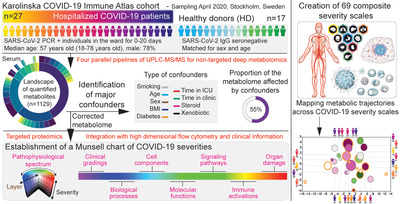
Results and discussion Massive remodeling of the circulating metabolome in COVID-19 and association with known confoundersWe performed a deep untargeted metabolomic profiling of serum from hospitalized moderate and severe COVID-19 patients (n = 27) and healthy donors (n = 17, matched for age and sex), using four parallel pipelines (Fig. 1a). This strategy allowed us to quantify more than 1100 metabolites from which 42% were lipids, 20% amino acids, and 20% xenobiotics (Fig. 1b and Supporting Information Table S1). In line with previous reports [10-12], 46%–75% of all metabolites, depending on the type, were significantly altered (FDR corrected p-value) in hospitalized COVID-19 patients as compared with healthy donors (Fig. 1c and Supporting Information Table S2). Detected compounds increased or decreased in most groups of metabolites, which was not surprising, given the complex therapeutic schemes of the patients. On the other hand, most xenobiotic substances measured were present at considerably higher concentrations in COVID-19 patients as compared with controls (Fig. 1c and Supporting Information Table S2). Since this suggested that a part of the observed differences between patients and controls were not directly caused by the infection and the ensuing inflammatory immune response, but rather by external factors such as administered drugs, we next analyzed the relationship between xenobiotic metabolites and other metabolites. Indeed, 486 highly significant interdependences were found (Fig. 1d and Supporting Information Table S3) primarily linked to 106 specific xenobiotic compounds co-varying with another 186 metabolites (Supporting Information Table S4).

Remodeling of all metabolic compartments in COVID-19 and identification of confounding factors. (a) Study design. Cohort and key characteristics are described as well as the pipeline of ultraperformance liquid chromatography (UPLC)-tandem mass spectrometry (MS/MS) electrospray ionization (ESI) used for metabolomics characterization of serum. (b) Landscape of quantified metabolites. Relative proportions of superpathways and the most common sub-pathways are depicted (number of metabolites identified). (c) Differences in metabolite concentrations between healthy donors (HD, n = 17) and hospitalized COVID-19 patients (n = 27). Each dot represents a single metabolite, FDR, false discovery rate; circular diagrams depict in blue the proportion of each superpathway significantly affected (q < 0.05). (d) Schematic view of the identification of xenobiotic-metabolite interdependencies. R, Spearman coefficient of correlation, the thinner circular diagram depicts the number of highly significant correlations between all metabolites while the thicker circular diagrams show the main xenobiotic-metabolite interdependencies and the related superpathways involved. (e) Impact of confounding factors on the metabolic landscape in hospitalized COVID-19 patients. Differences in metabolite concentrations between patients with or without the indicated confounding factors (y-axis) and healthy donors (x-axis). Each dot represents a single metabolite. The number of most variable metabolites (>fold change of 2, blue) and most significant (FDR corrected “q,” or not “p”) are indicated above each graph. (f) Distinct and shared effects of confounders on the metabolic landscape in hospitalized COVID-19 patients. Bar graphs show the number and the composition (superpathways, colored in the bars) of the metabolites affected (most variable on the top, most significant on the bottom plot) by the corresponding confounders (color-coded) indicated on the left. Circular diagrams show the proportion of the metabolome affected by confounders (including all xenobiotics). Pie charts display the proportion of metabolites affected by one or several confounders with single or shared effects displayed inside the upsets plots with connected dots under the bars.
To further decipher variations in the circulating metabolome in hospitalized COVID-19 patients associated with other confounding factors, metabolome alterations were characterized in relation to comorbidities and patient management factors (Fig. 1e). In the here studied COVID-19 cohort, age, sex, body mass index, presence of diabetes, and smoking all affected the metabolome (Fig. 1e and Supporting Information Table S5). Similarly, days between admission to hospital (to a regular ward or intensive care unit) and blood sampling for serum metabolomics had an impact on the serum metabolome. However, the most dramatic effect on the metabolome was seen by corticosteroid treatment (administered to 14 of 27 patients) prior to sampling. When aggregating significantly different metabolites (Fig. 1e), between 36% and 55% of the total metabolome was influenced by the above-mentioned cofactors and all types of metabolites were affected (Fig. 1f). Most of the alterations observed were specific to a single cofactor and not shared between several confounders (Fig. 1f and Supporting Information Table S6). Altogether, this shows that the circulating metabolome was considerably altered in hospitalized COVID-19 patients but that up to half of these alterations could be attributed to the confounding factors tested.
Integrating correction for confounding factors into general metabolomic measurementBy excluding xenobiotics and metabolites affected by confounders, a corrected metabolome for hospitalized COVID-19 patients was defined (Supporting Information Table S7). To evaluate the impact of such correction for biological interpretation, three proof-of-concept case studies were performed (Fig. 2a–c).

SARS-CoV-2-specific metabolic imprint across COVID-19 severities and multidimensional map of subclinical phenotypes. (a) Phylogeny deduced from serum metabolic assessment of healthy donors (#1-17) and COVID-19 patients (#18-44) using the corrected and noncorrected metabolome. Euclidian distances are calculated in the multidimensional reduced metabolic space (all principal components) and clustered using Ward's method. (b) Analysis of metabolic switches across pseudotime of early COVID-19 using the corrected and noncorrected metabolome. Metabolite concentrations are compared using Mann–Whitney test between healthy donors (n = 17) and hospitalized serum PCR positive/IgG negative (n = 5) or serum PCR negative/IgG positive (n = 12) COVID-19 patients at the time of sampling. Pie charts show the overlap between the top 100 significant changes found using corrected and noncorrected metabolome. Waterfall plots display the magnitude of changes identified using the corrected metabolome. Each bar represents a single metabolite. (c) Identification of predictive models distinguishing COVID-19 developing fatal cases using the corrected and noncorrected metabolome. Results from the best identified models using logistic regressions (0.05 as significance for entry and stay in the model, Akaike information criterion [AIC]) run either with the corrected or the noncorrected metabolome. (d) Schematic view of the structure and content of the Munsell chart of COVID-19 severities containing 69 composite severity scales across layers of the pathophysiological spectrum. (e) Correlation-based metabolic trajectories across the 69 composite severity scales of the Munsell chart. R, Pearson's coefficient of correlation. Combined clustering (using Ward's method) of severity scales and metabolites based on the three highest and three lowest Pearson correlation coefficients identified for each scale. (f) Correlations between single metabolites and selected serum proteome-defined neurological severity scales. Indicated p-values shown for linear regression calculations. (g) Map of the pathophysiological landscape of COVID-19 based on principal component (PC) analysis.
First, a COVID-19 metabolic phylogeny was defined using multidimensional reduction to allow for interpretation of similarities and differences in the high-dimensional dataset. A noncorrected metabolome overestimated the Euclidean distance differences between healthy donors and hospitalized COVID-19 patients as well as impacted the detailed order of each patient (Fig. 2a).
Second, independent group comparisons are often used to reconstruct “pseudotime” and emphasize temporal transitions in cross-sectional designs. Here, we tested this approach to evaluate metabolic switches happening at the transitions between healthy and the initial infectious replicative phase (severe acute respiratory syndrome coronavirus-2 [SARS-CoV-2] serum viremia), and the subsequent seroconversion phase (anti-SARS-CoV-2 IgG positive). When analyzing these transitions using a noncorrected versus a corrected metabolome and selecting the top 100 most significant changes, only a minority of the identified metabolites overlapped (violet pie chart, Fig. 2b). Instead, these transitions were driven by xenobiotic metabolites and a distinct lipid profile for the noncorrected metabolome (Fig. 2b). To gain insight into these metabolic switches, a resource using the corrected metabolome describing sequential up and downregulation of metabolite types upon viral replicative and seroconversion transitions was generated (Supporting Information Table S8).
Third, multivariate regression analysis is often used to build predictions models and is a basis for machine learning algorithms. Thus, the approach to distinguish hospitalized COVID-19 patients with an in hospital fatal outcome compared to those that recovered was tested (Fig. 2c and Supporting Information Table S9). Xenobiotic metabolites drove the best identified model (4-methylcatechol sulfate, benzoate metabolism) and the perceived effect of a common metabolite (6-bromotryptophane, -144 vs. -197) for both models was impacted by correcting the metabolome. Although containing almost only half of the metabolites, a metabolome corrected for confounding factors might still provide relevant models to predict outcome (Fig. 2c and Supporting Information Table S9).
Metabolomic profiling of serum is a powerful tool to capture fine biological perturbations and provide mechanistic insights in multiorgan diseases [8, 9]. However, many confounders need to be considered to uncover relevant signatures [13]. Here, we estimate that a considerable fraction (one-third to half) of the detectable metabolome in hospitalized COVID-19 patients is affected by associated risk factors and patient management. Metabolomics is a sensitive approach that can be influenced by food consumption, physical activity, administered treatments, or comorbidities such as obesity and diabetes. All these not only differ in hospitalized COVID-19 patients compared with healthy donors, but also within COVID-19 patients with moderate to severe forms of disease. Consequently, prediction models using metabolomics might be influenced by such confounders since more severely ill patients will be mostly older males with comorbidities, immobilized for longer periods of time in the hospital while receiving parenteral nutrition and various drug treatments such as corticosteroids.
Furthermore, the confounders have a cumulative effect, meaning that correcting for only a few of them (such as age, sex, and body mass index) might not be sufficient. Thus, previous studies on the metabolomic profile of COVID-19 should be considered in this context. For instance, in one prior report, more than 80% of the severe COVID-19 patients studied had received corticosteroids and immunoglobulins while few of the nonsevere patients had been given a similar treatment [12]. Similarly, in a second study comparing the metabolome of mild and severe COVID-19 patients, underlying comorbidities were three times more common in the severe group and 60% of those patients had also received corticosteroids while none of the patients with mild disease had [10]. To both provide useful mechanistic insights and surrogate markers, we here provide a resource of a COVID-19 metabolic imprint corrected for these confounders.
In summary, correcting the metabolome for confounders in COVID-19 impacted a broad range of statistical models and is critical for biological interpretations.
Establishment of proteomic and flow cytometry-based severity scalesIn searching for metabolic fingerprints of key pathophysiological mechanisms in hospitalized COVID-19 patients, an independent proteomic dataset was generated on the same patients. Among 1472 serum proteins quantified, biological- and organ-specific signatures were defined based on pathway analysis and organ-specific gene expression patterns [14] (Supporting Information Tables S10 and S11 and Fig. S1). In addition, high-dimensional flow cytometry analysis was performed on matched peripheral blood immune cells to establish cell-specific immune activation signatures (Supporting Information Table S10). These signatures defined composite severity scales that were significantly altered in hospitalized COVID-19 patients as compared to healthy donors (Supporting Information Fig. S2). Overall, a Munsell chart of COVID-19 severities containing 69 composite scales allowing the dissection of specific aspects of a wide pathophysiological spectrum from clinical phenotypes to biological mechanisms was generated (Fig. 2d, Supporting Information Table S10 and Fig. S2).
Association between metabolic imprint in COVID-19 and CNS damageUsing correlation analysis between the corrected metabolome and the generated severity scales, common and distinct metabolic trajectories across layers of the pathophysiological spectrum were identified (Fig. 2e and Supporting Information Fig. 3). This provided a metabolic atlas to deconvolute pathophysiological events in COVID-19. Some trajectories, such as group 1, were dominated by a narrow modification of the lipid profile with acyl-carnitin derivatives representing four out of 12 metabolites of the group, associating with brain and carbonate dehydratase severity scales (cluster II). Others retained a much diverse composition (such as group 3) and associated with clinical scales of severity (cluster V). The largest common metabolic imprint was detected in group 6 that also contained 6-acyl-carnitine derivatives positively correlated with a large range of biological, cellular, molecular, and organ damage severities including lung damage (cluster I). On the other hand, metabolic group 6 presented negative correlations with immune-related severity scales (clusters VI, VII, IX, and X), suggesting a distinct immune-metabolic trajectory. Finally, liver and kidney damage (cluster IV) shared similar metabolic signatures distinct from intestine and brain damage (cluster II) but both showing positive correlations with the acyl-carnitine derivatives from the group 1. Across this metabolic atlas of severity scales, using MetaboAnalyst 5.0 identified ranges of single nucleotide polymorphisms associated with metabolic traits including carnitine derivatives (Supporting Information Table S12) including rs6886262 targeting actin-binding LIM protein 3 (ABLIM) expressed in muscle, liver, parathyroid gland, and brain with potential functional relevance [15]. Moreover, the concentration of palmitoylcarnitine was correlated with severity scales of axon, glutamatergic synapse, and brain damage (Fig. 2e and Supporting Information Fig. S3), which was of particular interest given that single nucleotide polymorphisms in the gene encoding carnitine palmitoyltransferase II were previously described as a predisposing factor for encephalopathy in influenza and other infections [16]. As an example, we further illustrated how metabolic trajectories reflecting neurological scale severities could identify potential surrogate markers to assess subclinical pathological mechanisms in patients (Fig. 2f).
Beyond linking the metabolic imprint in COVID-19 to CNS damage, these results also raised the possibility that certain metabolic features associated with COVID-19 pathogenesis are genetically influenced.
Integrated multidimensional analysis of proteomic and immune-related severity scales in COVID-19To gain a deeper insight into common and distinct pathophysiological mechanisms in COVID-19, a dimensionality reduction based on all biological severity scales was performed (Fig. 2g and Supporting Information Table S13). Principal component 1 (PC1) identified a major role for hormone response and neuronal death in driving the heterogeneity of patients, although most scales contributed to this component, thus identifying the common global pathway of alterations in hospitalized COVID-19 patients. Other components offered possibilities to identify distinct independent phenotypes within the multidimensional pathobiological continuum. First, activation of the monocyte compartment appeared confined to different patients as those presenting with elevated carbonate dehydratase and BM signatures (PC2). Second, robust neutrophil activation and parathyroid signatures were opposite to increased platelet and neurotrophin signatures (PC3). Third, neuroinflammatory response together with regulation of IL6 production was opposed to activation of non-naïve CD4 T cell, mucosal invariant associated T cells, and intestinal damage (PC4). Thus, we here mapped the pathophysiological landscape of COVID-19 identifying common and distinct phenotypes that reveals a role of the neuroendocrine system.
Although the immunopathology of COVID-19 has been relatively well characterized [17], its immunometabolism remains largely unexplored despite both a high pathophysiological and clinical relevance [18, 19]. We here show that metabolite concentrations correlating with immune activation are distinct from those reflecting clinical gradings and organ damage. As an exception, we found that the metabolic trajectory correlating with eosinophils activation clustered tightly with severity scales of multiple pathways and organ damage (including lung, cluster I). Also, the activation of type 2 innate lymphoid cells (cluster IX) appeared to be linked to relatively specific features, such as positive correlations with the level of bilirubin derivatives (group 3). Whether these metabolic signatures are the cause or consequence of the immune activation and organs damage cannot be assessed in our cross-sectional setting. However, speculatively, these data could indicate a peculiar role for type-2 immunity involving type 2 innate lymphoid cells and eosinophils [20] in COVID-19.
Limitations of this study are important to consider for interpretation of the current results and design of future studies. First, the current sample size precludes definitive conclusions to be drawn and raises the need for validation in larger cohorts and longitudinal settings. In addition, the establishment of a corrected metabolome, although key to unravel COVID-19 specific features, is likely to mask clinically and biologically relevant results concerning specific subgroups of patients such as individuals presenting with one or a specific combination of comorbidities. Finally, the specificity and confidence in the pathogenic events detected need to be carefully considered. We have relied on groups of proteins, datasets from the Human Protein Atlas, and pathway analysis with the aim to map distinct human organs or biologically relevant mechanisms with high specificity probably ensuring high accuracy and biological relevance. However, we anticipate detecting subclinical pathogenic events that might be difficult to validate experimentally in local tissues in humans.
Concluding remarksBeside the immune system, we mapped the relationship in-between COVID-19 phenotypes that suggested a contribution of the neuroendocrine system in driving patient heterogeneity. As many hospitalized COVID-19 patients present with a large spectrum of neurological symptoms, including acute encephalopathy, there is an urgent need to better understand and detect pathophysiological events related to the CNS [3, 21]. Current knowledge mainly stems from autopsy studies of fatal COVID-19 cases and suggests that SARS-CoV-2 can penetrate the brain where it has been associated described with neuroinflammation, microthrombus, and tissue damage [1, 2, 7]. Our data suggest that diverse forms of neuropathies might be present since neuron death associated with viral entry into host cells, response to hormone and hypoxia, but did not co-occur with neuroinflammation, platelet activation, or neurotrophin signaling. Thus, these might either be temporally distinct events or distinct pathophysiological pathways. Since large-scale personalized proteomics is not yet a realistic approach in clinical diagnostics, the assessment of a single (or a few) metabolite as surrogate marker might be an alternative cost-effective approach rapidly implementable in clinical routine. In such a context, the provided scales and the associated correlated metabolites might allow to detect various immune activators, poor metabolic trajectories, organ damage before clinical deterioration, or activation of various signaling pathways with implications for patient monitoring and targeted therapies with high specificity. Overall, we here provide a metabolic atlas across COVID-19 severities and an insight into system level pathogenesis, including CNS damage.
Materials and methods Characteristics of patients and controlsSARS-CoV-2 RNA+ patients with moderate (n = 10) or severe (n = 17) COVID-19 were recruited to the study and sampled 0–8 days after being admitted to hospital. Healthy controls were SARS-CoV-2 IgG seronegative at time of inclusion. The study was approved by the Swedish Ethical Review Authority and all patients gave informed consent. More details are available on www.covid19cellatlas.com.
Sample preparation and deep metabolomicsAll serum samples were processed within 4 h from them being taken, frozen, and maintained at –80°C until processed. Samples were prepared using the automated MicroLab STAR® system (Hamilton Company). The resulting extract was divided into fractions: two for analysis by two separate reverse phase/UPLC-MS/MS methods with positive ion mode electrospray ionization (ESI), one for analysis by reverse phase/UPLC-MS/MS with negative ion mode ESI, and one for analysis by HILIC/UPLC-MS/MS with negative ion mode ESI.
Defining a corrected metabolomeTo distinguish the metabolome specifically associated with COVID-19 rather than with clinical risk factors and/or clinical management, metabolites that varied according to a range of risk factors and management criteria were censored from the global metabolome.
Targeted proteomicSerum proteomics was performed using the Proximity Extension Assay technology (Olink Explore 1536) were 1472 proteins and 48 controls were measured in each sample.
Pathogenic biological events and tissue-specific damageOrgan damage scores were defined based on serum proteins significantly different in COVID-19 that also mapped specifically to an individual human tissue (Supporting Information Fig. S1 and Table S10). Pathway analysis was based on several sets of serum proteins significantly associated with specific biological events that at the same time were significantly altered in COVID-19 patients (Supporting Information Fig. S2).
Flow cytometryPBMC were stained fresh for flow cytometry and acquired on a BD LSR Symphony. More details on flow cytometry panels used are available on https://covid19cellatlas.com. Absolute counts from the different samples were obtained using BD Trucount™ tubes.
StatisticsIf not specified, Mann–Whitney U-test and Spearman’ rank coefficient were used to analyze differences between groups and correlations, respectively. False discovery rate was applied to correct for multiple comparisons when mentioned. Clustering was based on Euclidian distances and Wards’ method. More details on the statistical methods are provided in the Supporting Information.
AcknowledgementsWe express our gratitude to all patients, health care personnel, study coordinators, administrators, and laboratory managers involved in this work.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
FundingThe work has been supported by Knut and Alice Wallenberg Foundation, Nordstjernan AB, European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 948692), the Swedish Research Council, the Swedish Cancer Society, the Swedish Foundation for Strategic Research, and the Center for Innovative Medicine at Karolinska Institutet, Region Stockholm.
Author contributionsConceptualization and methodology: MC and NKB. Analysis and manuscript drafting: MC and NKB. Experimental data contribution: MC, BS, AP, PC, JRM, MA, MB, BJC, MD, IF, JBG, SGR, LH, JK, EK, EK, ML, JM, CM, ANT, TP, APP, ORB, JKS, JTS, TS, MS, RV, and HGL. Clinical data contribution: OR, LIE, SA, KS, and HGL. Revising the final manuscript: all authors.
Study groupThe members of the Karolinska COVID-19 Study Group are Mira Akber, Soo Aleman, Lena Berglin, Helena Bergsten, Niklas K. Björkström, Susanna Brighenti, Demi Brownlie, Marcus Buggert, Marta Butrym, Benedict Chambers, Puran Chen, Martin Cornillet, Jonathan Grip, Angelica Cuapio Gomez, Lena Dillner, Therese Djärv, Jean-Baptiste Gorin, Isabel Diaz Lozano, Majda Dzidic, Johanna Emgård, Lars I. Eriksson, Malin Flodström-Tullberg, Hedvig Glans, Sara Gredmark-Russ, Quirin Hammer, Alvaro Haroun-Izquierdo, Elisabeth Henriksson, Laura Hertwig, Sadaf Kalsum, Tobias Kamann, Jonas Klingström, Efthymia Kokkinou, Egle Kvedaraite, Hans-Gustaf Ljunggren, Marco Giulio Loreti, Magda Lourda, Kimia T Maleki, Karl-Johan Malmberg, Nicole Marquardt, Christopher Maucourant, Jakob Michaelsson, Jenny Mjösberg, Kirsten Moll, Jagadeeswara Rao Muvva, Johan Mårtensson, Pontus Nauclér, Anna Norrby-Teglund, Annika Olsson, Laura M Palma Medina, Tiphaine Parrot, Björn P Persson, André Perez-Potti, Lena Radler, Dorota Religa, Emma Ringqvist, Olga Rivera-Ballesteros, Olav Rooyackers, Johan K. Sandberg, John Tyler Sandberg, Takuya Sekine, Ebba Sohlberg, Tea Soini, Kristoffer Strålin, Benedikt Strunz, Anders Sönnerborg, Mattias Svensson, Janne Tynell, Renata Varnaite, Andreas Von Kries, Christian Unge, and David Wulliman.
留言 (0)