
記住我







Approaches to enhance T cell reactivity have yielded impressive results in the treatment of cancer. In particular, the use of CD19-targeted chimeric antigen receptor (CAR) T cells has revolutionized the treatment of B-cell leukemias, whilst function-blocking antibodies targeting immune checkpoint receptors such as programmed cell death protein 1 (PD-1) or cytotoxic T lymphocyte-associated protein 4 (CTLA-4) have proven effective in several cancer types. Nonetheless the success of CAR-T cell therapy in hematological conditions has not, as yet, been replicated in solid tumors, whilst even in immunotherapy-sensitive cancers such as melanoma, approximately 50% of patients fail to benefit from the use of immune checkpoint inhibitors. Therefore, there remains an urgent clinical need to identify and validate additional cellular targets to revive T-cell responses in cancer. Whilst much work has focused on the role of cell surface co-inhibitory receptors, less is understood of the intracellular checkpoints that limit T-cell anti-cancer immunity. To begin to address this knowledge gap, a recently published study by Wiede and colleagues identified phosphotyrosine phosphatase 1B (PTP1B) as a key negative regulator of T-cell responses in cancer and provided compelling evidence that inhibition of PTP1B function represents a rational approach to improving T cell-mediated cancer immunotherapies.1
PTP1B is a widely expressed protein and has key roles in the regulation of metabolic signaling.2 Furthermore, previous studies have described tumor cell-intrinsic roles for PTP1B in cancer progression (reviewed in Yip et al.3). Wiede et al. have set out to determine whether PTP1B regulates immune responses to cancer. Using whole body knockout mice and an ovalbumin-expressing orthotopic model of mammary cell cancer (AT3-OVA), the authors determined that tumor growth was reduced in the absence of PTP1B expression. This effect was associated with enhanced numbers of tumor infiltrating leukocytes (TILs) and was recapitulated in chimeric mice in which PTP1B expression was absent in bone marrow-derived cells but expressed in stromal compartments. Importantly, T cell-specific deletion of PTP1B, using Lck-driven expression of Cre recombinase, resulted in enhanced anti-tumor responses and suppression of AT3-OVA tumor growth, confirming a T cell-intrinsic role for PTP1B as an intracellular immune checkpoint. These results add to a body of work that has determined roles for phosphotyrosine phosphatase non-receptor type (PTPN) proteins as key regulators of anti-tumor T cell immunity. Thus, additional members of the PTPN family have previously been implicated as intracellular checkpoints of T-cell responses to cancer, including the closely related PTPN24 and additional family members PTPN65 and PTPN22.6
T-cell exhaustion within the tumor microenvironment is defined by a progressive loss of effector function and metabolic fitness and is associated with elevated expression of immune checkpoint receptors such as PD-1, CTLA-4, lymphocyte activation gene 3 (LAG-3) and T-cell immunoglobulin and mucin domain containing-3 (TIM-3).7 The high expression levels of immune checkpoint receptors on exhausted T cells are thought to be a consequence of chronic inflammation and/or antigenic stimulation. Whether expression of intracellular checkpoints is similarly modulated in TILs is less well understood. In this regard, Wiede and colleagues demonstrated that PTP1B expression was elevated approximately two-fold in effector/memory phenotype tumor-infiltrating CD8+ T cells compared with comparably activated splenic T cells.1 Interestingly, heterozygous deletion of PTP1B in T cells was sufficient to enhance the control of tumor growth, suggesting that the two-fold increase in PTP1B expression shown for TILs is likely to be functionally relevant. Analysis of published single cell RNA sequencing of human TILs linked high PTP1B expression with effector or exhausted T-cell gene expression profiles,1 suggesting that PTP1B might play a similar role in limiting human T-cell responses in cancer. The finding that PTP1B is upregulated selectively in TILs but not in activated splenic T cells is intriguing. The signal(s) within the tumor microenvironment that drive elevated PTP1B expression were not assessed but might include chronic inflammation mediated by TNF or other inflammatory cytokines, chronic TCR stimulation that is known to drive expression of co-inhibitory receptors or, potentially, immuno-inhibitory cytokines or metabolites. Future work is required to distinguish between these possibilities.
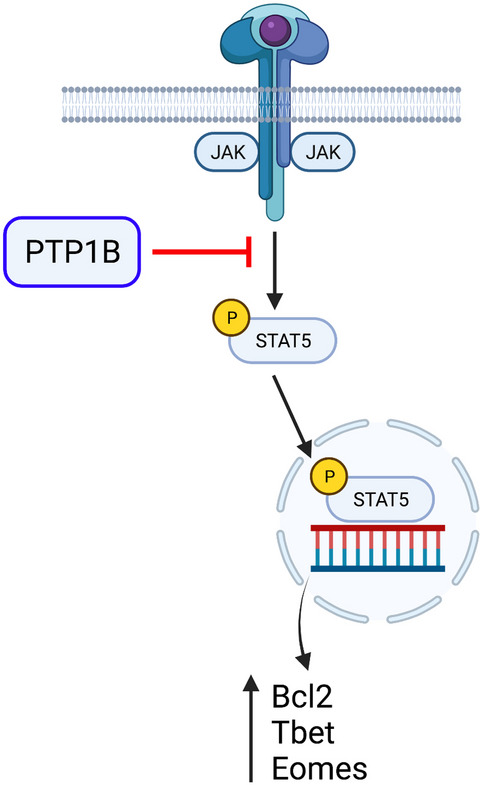
To define the mechanisms underpinning the effects of PTP1B deletion, Wiede et al. assessed the regulation of T-cell receptor (TCR) and cytokine signaling in knockout T cells. Whereas signaling events following TCR triggering were largely unaffected, the levels of basal and inducible phosphorylation of signal transducer and activator of transcription 5 (STAT5) were elevated in PTP1B-deficient T cells. STAT5 is a key transcription factor downstream of the interleuin (IL-2) receptor8 and other common gamma-chain cytokines, and elevated STAT5 phosphorylation in PTP1B-deficient T cells was associated with increased expression of known downstream targets such as the anti-apoptotic factor BCL-2 and key transcription factors Tbet and eomesodermin (Figure 1). Importantly, heterozygous deletion of STAT5 reversed the enhanced activation of PTP1B-knockout T cells and the repression of tumor growth in T cell-specific PTP1B-deficient mice. These data indicated that regulation of cytokine signaling is likely the key target of PTP1B. Of note, recombinant IL-2 was one of the earliest immunotherapies that demonstrated any degree of clinical efficacy in human cancers, although its use has largely been superseded by the use of immune checkpoint inhibitors.9 The new studies by Wiede and colleagues demonstrate that modulation of cytokine signaling pathways remains a viable target to revive T-cell responses in cancer.

PTP1B acts as an intracellular checkpoint for T cell anti-cancer responses by blocking cytokine receptor signaling. Common gamma chain cytokine receptor signaling induces activation of JAK and Tyk kinases and subsequent phosphorylation of transcription factor STAT5. Phosphorylated STAT5 promotes T-cell activation via transcription of key target genes including BCL2, Tbet and Eomesodermin (Eomes). Wiede and colleagues demonstrated that PTP1B acts to dampen cytokine signaling by attenuating JAK, Tyk2 and STAT5 phosphorylation. Furthermore, elevated STAT5 activation contributes to enhanced anti-tumor T-cell responses following genetic or pharmacological inhibition of PTP1B. The figure was created with BioRender.com.
Current immunotherapies can be broadly categorized as either cell-based therapies (e.g. CAR-T or other adoptive T cell transfer-based therapies) or targeted therapies using biologicals or small molecule inhibitors (e.g. immune checkpoint inhibitors). Impressively, Wiede and colleagues determined that modulating PTP1B activity has potential for use in both cell-based and targeted immunotherapies. Strikingly, the authors showed that deletion of PTP1B enhanced CAR-T cell function in a mouse model of triple negative breast cancer, a solid tumor type currently lacking effective immunotherapies. Importantly, as well as demonstrating effective anti-tumor responses, PTP1B-deficient CAR-T cells did not instigate any signs of systemic inflammation or substantial morbidity, a key limitation of CAR-T cell use in cancer patients. Furthermore, the researchers also provided evidence that enhancement of T-cell function in cancer can be achieved using a PTP1B small molecule inhibitor MSI-1436. Combinations of MSI-1436 with anti-PD-1 antibody provided the greatest protection from tumour growth in mice, indicating the potential for combining PTP1B inhibition with existing immunotherapies to achieve superior clinical outcomes.
The finding that pharmacological inhibition of PTP1B can be effectively combined with PD-1 inhibitors in mice highlights the importance of both the well-described immune checkpoint receptors and intracellular signaling checkpoints in the regulation of anti-tumor T-cell responses. It is also worth emphasizing that the transmission of inhibitory PD-1 signaling itself involves the recruitment and activity of tyrosine phosphatases PTPN6 (SHP-1) and PTPN11 (SHP-2).10 Importantly, PD-1 acts primarily to block TCR-driven signals, whereas the results from the current work demonstrate that PTP1B deletion or inhibition impacts primarily upon cytokine signaling. It will be interesting to determine whether PTP1B inhibition might prove an effective alternative to PD-1 or CTLA-4 blockade in immunotherapy-resistant cancer types as well as building upon the finding that these approaches can be combined with increased efficacy in immunotherapy-sensitive cancers.
To advance this work, a key next step will be to further define the impact of PTP1B deletion in CAR-T and conventional TCR-expressing T cells in humans. Initial studies carried out by Wiede and colleagues in the current work are encouraging; CRISPR-mediated knockout of PTP1B in human T cells recapitulated some of the key phenotypes demonstrated for mouse cells.1 Notwithstanding these challenges, the work by Wiede and colleagues provides a clear path for the development of PTP1B-targeted therapies that have the potential to improve the efficacy of immune responses and to provide benefit to cancer patients.
Robert Salmond: Conceptualization; Writing – original draft; Writing – review & editing.
留言 (0)