
記住我
Inflammatory myofibroblastic tumour (IMT) of the lung is a rare finding, comprising less than 1% of all surgically resected lung lesions. Nevertheless, in paediatric ages, it is a common primary lung tumour and is usually found incidentally.1-7
IMT of the lung can be presented as an isolated mass but it can also be locally invasive.1, 8-10 It is still a dilemma for these tumours, whether they represent a pure inflammatory process or a low-grade malignancy with a dominant inflammatory response. Here, we describe the management of a case and also a review of the literature.
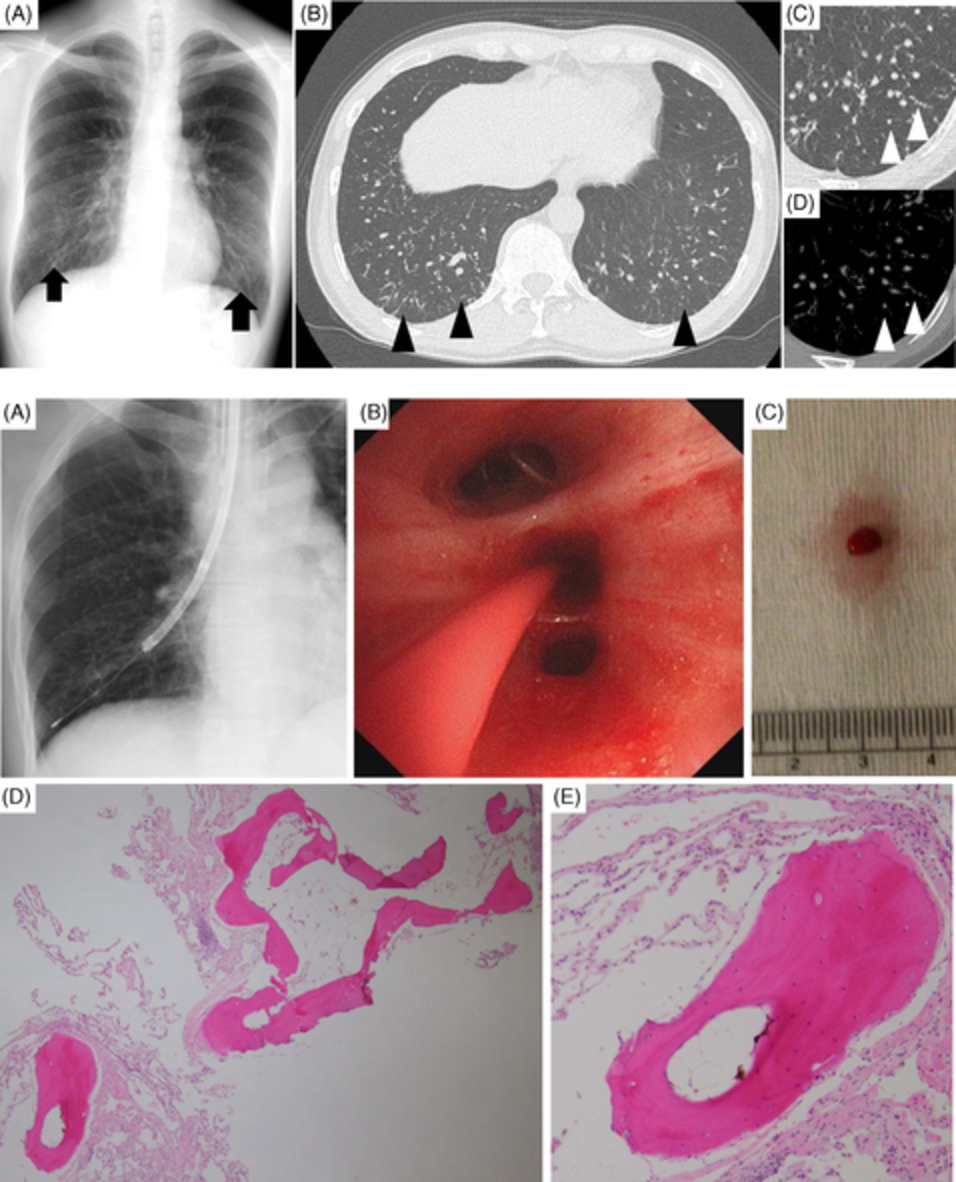
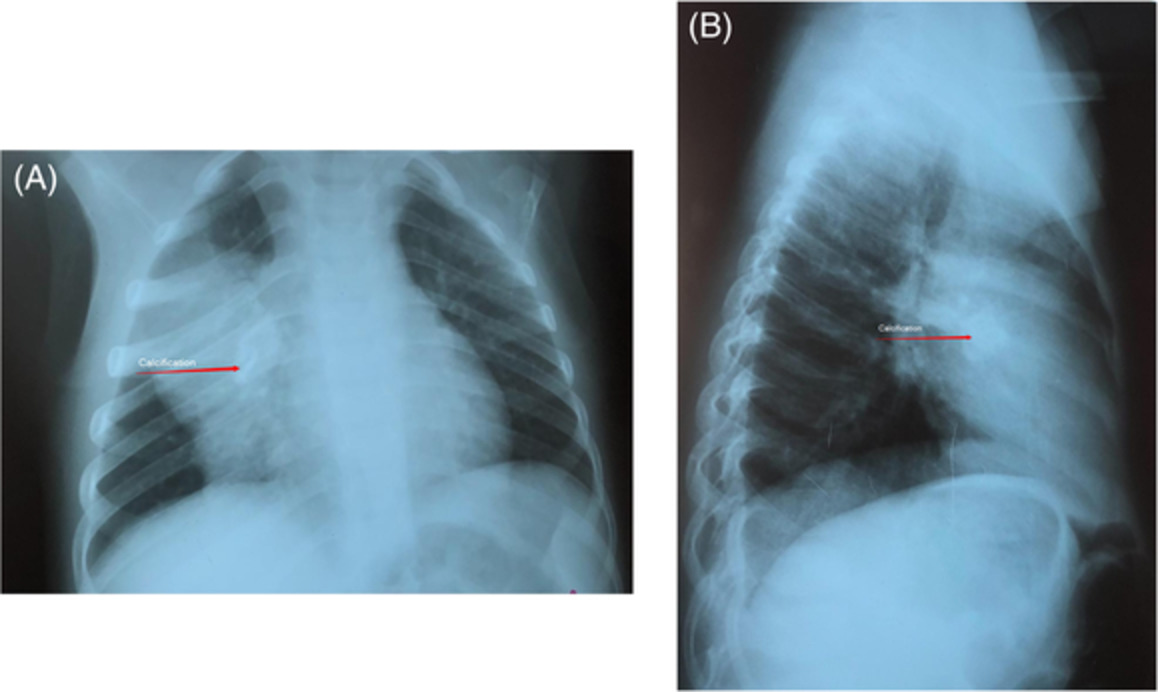
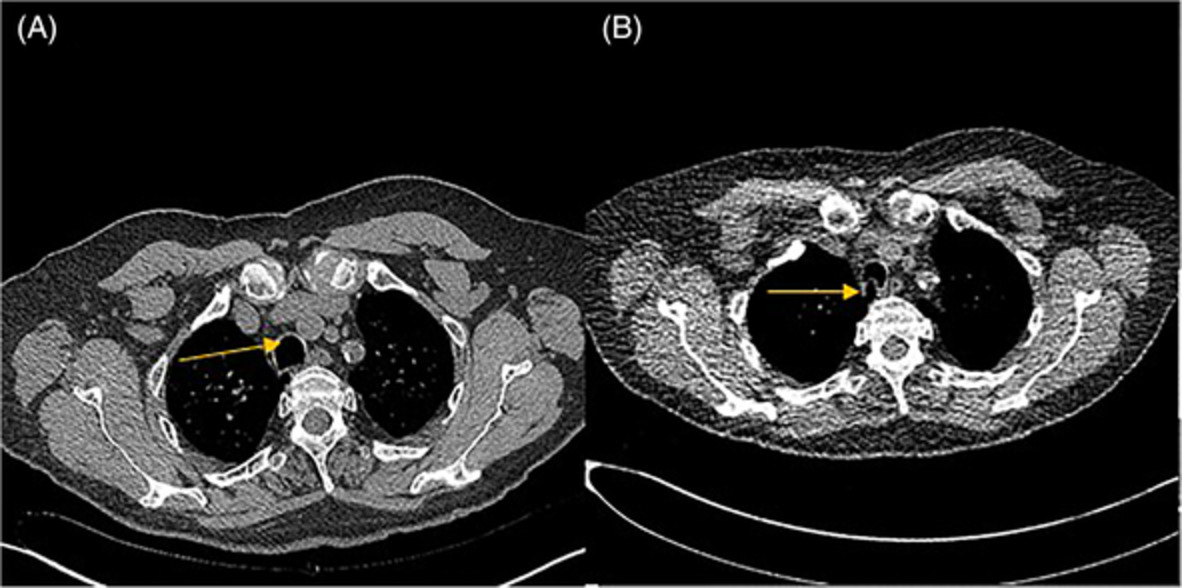
CASE REPORTA 4-year-old girl was referred to our tertiary hospital for the specialist evaluation of a non-specific cough, which was present for 6 weeks, associated with a right pulmonary mass. The physical examination was normal; on auscultation, diminished breath sounds were present on the right side of the lung. The medical history and growth were unremarkable. Chest radiograph revealed a mass located in the right upper lobe and the middle lobe of the right lung, with a central area of calcification (Figure 1). Chest computed tomography (CT) scan confirmed the chest radiograph findings; a solid, well-contoured, heterogeneous, mass was noted in the right upper lobe and middle lobe of the lung with an area of central calcification (Figure 2).

(A) Thorax radiography of the myofibroblastic tumour. (B) Lateral radiography of thorax shows myofibroblastic tumour of the lung

Computed tomography of the myofibroblastic tumour of the lung
No lymphadenopathy was detected. Microscopy, culture and cytology of the sputum were unremarkable.
The erythrocyte sedimentation rate was 10, haemoglobin was 11.6 g/dl and the leucocyte count was 8.8 × 109/L. The other serum haematological and biochemical results were normal. The serology of Echinococcus and Mantoux test were negative.
The patient did not respond to antibiotics; therefore, surgical removal of the mass was performed. Thoracotomy performed on the right side showed a lesion in the lung parenchyma, extended to the upper and middle lobes. The differential diagnosis of congenital lesions of the lung was made. As there was no success with fine-needle aspiration in several cases, we decided to perform surgical resection.
The lesion was resected and lobectomy of both upper lobe and middle lobe was also performed. No associated lymphadenopathy was noted.
Macroscopically, a well-circumscribed mass measuring 5.5 × 5.5 × 4 cm was present. The excised tumour had an osseous centre measuring 2 × 1 × 1 cm. From the histological point of view, the mass consisted of disorganization of the normal bronchoalveolar parenchyma, myofibroblastic cells and inflammatory cell infiltrates, such as lymphocytes, neutrophils, eosinophils and histiocytes. In the centre of the lesion, a large ossification was found.
Immunohistochemistry revealed calponin antigens, SMA, desmin, P53, bcL2, CK, HMW, and, in several myofibroblastic cells, expression of ALK-1. It was negative for beta-catenin, cyclin D1, Myo-D1, S100p, CD34, CD99 and CD117.
The histological and immunohistochemistry characteristics were compatible with an inflammatory myofibroblastic pseudotumor.
The post-operative course was uneventful, and the patient was discharged home after 2 weeks of surgery. The patient re-presented to the hospital 1 year after surgery. The child had normal daily activities, normal chest x-ray and no recurrences.
DISCUSSIONIMT is a rare tumour of the lungs, which was first described in 1939.8 They can grow to the lung, the brain, skin, soft tissues, liver and so on.8-15 They develop mostly in paediatric ages.2-7 The aetiology and pathogenesis are not fully understood. Some hypotheses support an abnormal immunological response towards viruses or foreign antigens; herpes type 8 virus and Epstein–Barr are mostly accused.16-18 Immunohistochemical results of IMT show the presence of polyclonal plasma cells, which support the fact that IMT is an inflammatory process.17, 18 Based on histopathological data, the other hypothesis presents IMT as low-grade neoplasms with a superimposed inflammation.19
The myofibroblast is recognized as the principal cell type of IMTs.20 Because of variable histology results, several synonyms are used for these masses, such as: plasma cell granuloma, fibrous histiocytoma, inflammatory pseudo-tumour, fibroxanthoma and xanthogranuloma.
IMTs are usually benign tumours, mainly occurring in paediatric ages. They are one of the most common lung tumours in patients younger than 16 years.1-7 Recurrence and/or malignant transformation of myofibroblastic tumour is a rare phenomenon. Markers for the neoplasia development include chromosome 2p23 reordering and ALK-1 expression.21 Coffin et al.17 showed that ALK-1 may be a marker of recidivism, but not a marker of malignant transformation. In the study by Chun et al.,18 the authors found a better prognosis in patients who tested positive for ALK-1. Meanwhile, recurrences may be attributed to incomplete resection of the mass.
These lesions have no gender difference, and represent less than 1% of lung tumours.1-7 Most of the cases have no symptoms and are discovered incidentally by chest radiograph.17
Physical examination and laboratory tests demonstrated no specific findings. The chest radiograph usually demonstrates a mass of 1–10 cm in diameter.23 Multiple nodules, calcifications and invasive disease have been reported occasionally.24 The diagnosis cannot be relied on chest CT and/or bronchoscopy and fine-needle aspiration biopsy. Surgery is recommended for diagnostic and therapeutic reasons.25, 26
Macroscopically, IMTs are firm yellow-white masses, have clear borders and may also have calcifications. They are usually located in the parenchyma, and sometimes may be located as endobronchial masses.
Microscopically, the lesions have fibroblasts, myofibroblasts, lymphocytes, plasma cells, histiocytes, neutrophils and eosinophils.20 Immunohistochemistry demonstrates the predominance of immunoglobulin G.17, 18
The diagnosis should be based on careful histological examination and even immunohistochemistry stains several times.20, 27 If possible, resection of the whole mass is recommended. To guide the extent of excision, frozen sections obtained at the time of operation time can be helpful, generally minimized to preserve lung function.28
The differential diagnosis includes pseudolymphoma, lymphosarcoma and fibrous histiocytoma.18, 20, 27, 29
Surgical resection of the whole mass is recommended, in order to exclude malignancy and completely treat the disease.18, 24, 26, 30 In cases when complete surgical resection is not possible, in multifocal disease or if recurrences occur, treatment options are radiotherapy, corticosteroids, chemotherapy and competitive inhibitors of ALK tyrosine kinase.3, 18, 31
The prognosis of myofibroblastic tumour is very good after total surgical removal, and the chances for recurrence and malignant transformation are low. Long-term follow-up is recommended after surgery.23
Clinicians need to be aware about the different clinical presentations of lung IMTs, which are rare, usually asymptomatic and may present as a pulmonary mass with well-defined borders that may resemble cancer. The best method to diagnose and treat them is surgical excision. As preoperative tests are not diagnostic, tumour's excision is necessary to exclude malignancy. When possible, total excision of the mass is recommended in order to prevent recurrence of the tumour.
AUTHOR CONTRIBUTIONSonila Boriçi: conception or design of the work; acquisition, analysis or interpretation of the data; drafting the work or revising it critically for important intellectual content; and final approval of the version to be published. Marjeta Tanka: drafting the work or revising it critically for important intellectual content. Luljeta Serbo: drafting the work or revising it critically for important intellectual content.
ETHICS STATEMENTThe authors declare that appropriate written informed consent was obtained for the publication of this manuscript and accompanying images.
留言 (0)