
記住我
Asphyxiating thoracic dystrophy (ATD) is a rare recessively inherited disorder that is most commonly detected in infancy and is often fatal. In this report, we describe an adult patient with his first presentation of respiratory failure and a presumptive diagnosis of ATD.
CASE REPORTA 57-year-old male of Southern Italian origin presented to hospital with a 6-month history of progressive dyspnoea and declining exercise tolerance. His medications at the time of admission were candesartan for hypertension, as well as regular inhaled budesonide/formoterol, and as required inhaled salbutamol that had been recently commenced by the patient's general practitioner for suspected asthma as the cause for these symptoms. He had required two previous ophthalmological procedures in early adulthood and remained vision impaired with retinal disease. He denied other significant medical or surgical history. The patient was a lifelong non-smoker and had not used e-cigarettes or any other inhaled agent.
In the 6 months prior to presentation, the patient had noted progressively worsening dyspnoea and exercise intolerance, acutely deteriorating preceding his admission to hospital. Prior to this, the patient denied any significant respiratory symptoms and was unrestricted in his exercise tolerance. These symptoms had initially been attributed to smoke exposure from recent Australian bushfires, and inhaled bronchodilators had been commenced with little improvement. There were no symptoms at the time of presentation to suggest a respiratory infection.
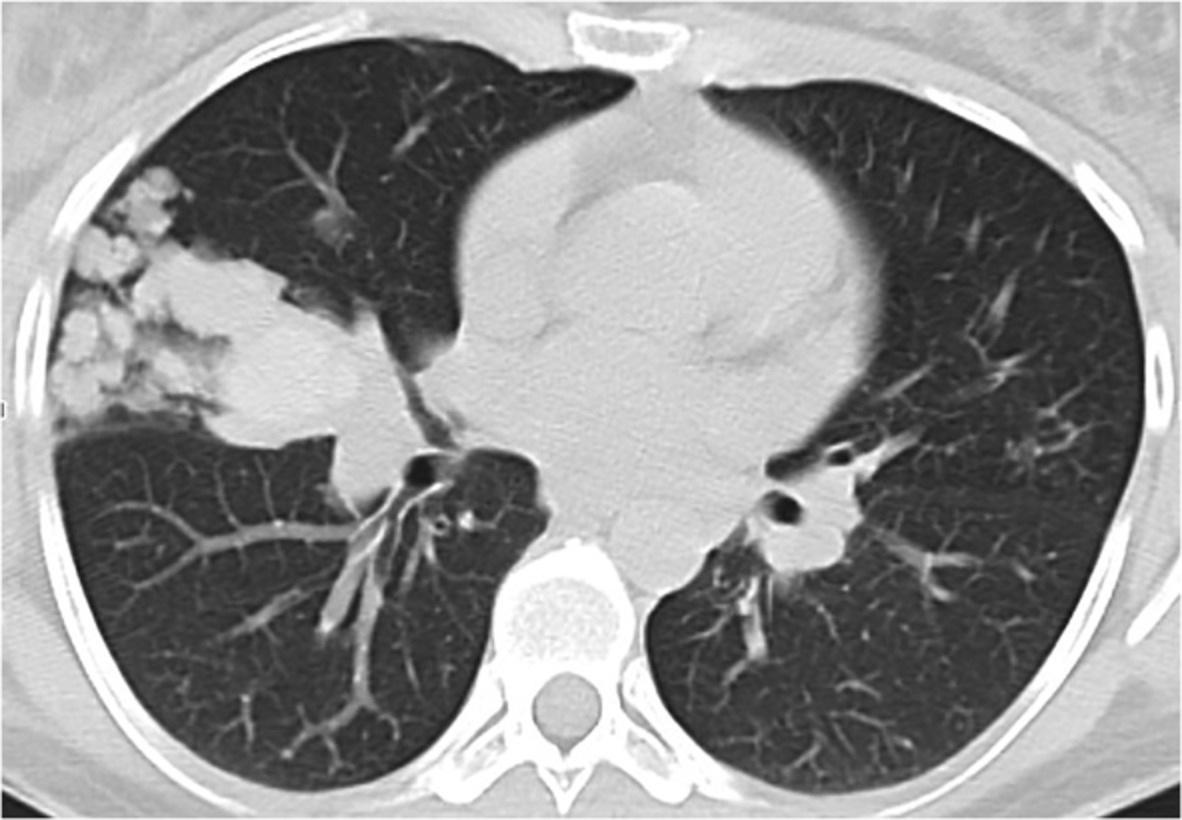
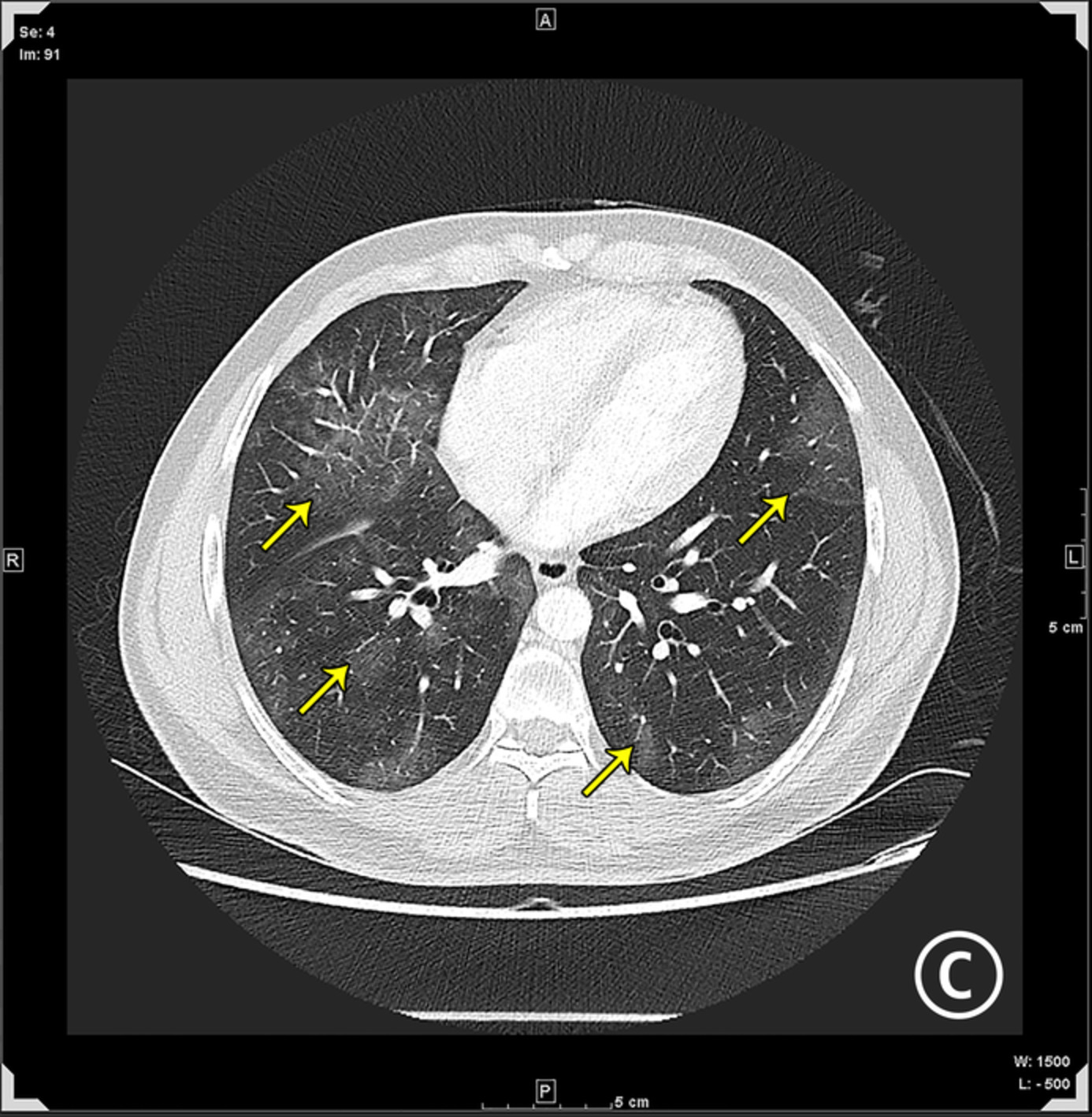
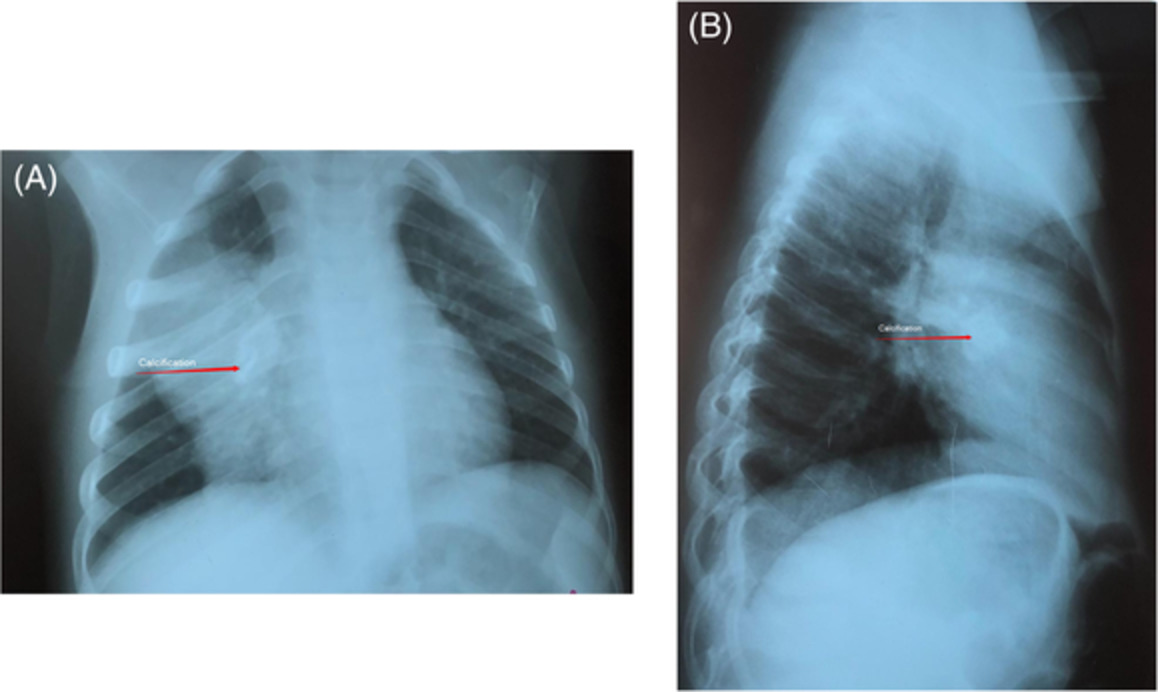
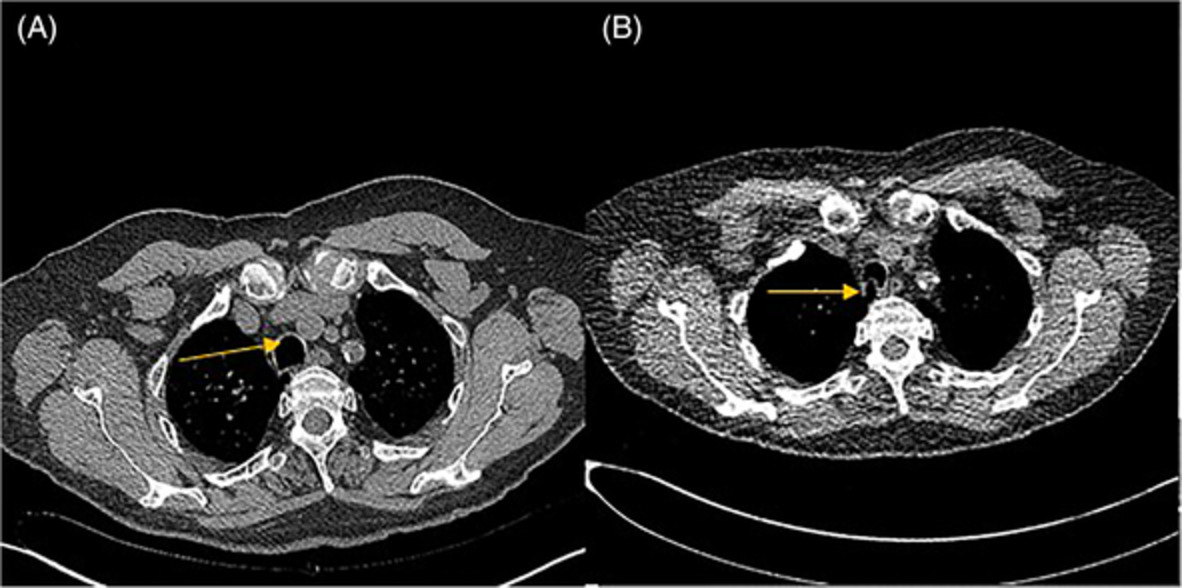
Clinical examination at presentation revealed a man of short stature (142 cm) with shortened limbs but otherwise normal digits. The chest was dysmorphic, with a bell-shaped chest and abdominal configuration, and marked symmetrically reduced chest expansion (Figures 1 and 2). There were no dysmorphic facial features and the patient appeared to be of at least average intelligence. Auscultation of the chest demonstrated very occasional basal crackles. Examination of other systems was largely unremarkable. Oxygen saturation at rest was maintained between 86% and 90% on room air. On mobilization, however, the patient was noted to become increasingly dyspnoeic, with peripheral oxygen saturation falling to 78% on room air. Chest computed tomography demonstrated marked chest wall deformity with a narrowed thorax, prominent rib shortening and profound reduction in size of the thoracic cavity, suggestive of an underlying chondrodysplasia (Figure 2). There was no evidence of pulmonary oedema or infection on imaging.

Anterior and lateral view of the patient, demonstrating characteristic dysmorphic ‘bell-shaped’ chest and abdomen, clavicular attenuation and limb shortening and thickening

Axial and coronal computed tomography view demonstrating the abnormal chest and abdominal configuration, with prominent rib shortening, thoracic narrowing and profound reduction in the size of the thoracic cavity
On further history, the patient described a relatively unremarkable childhood and adolescence, completing high school and working in retail since this time in a largely sedentary role. He recalled frequent visits to an ophthalmologist, but otherwise had surprisingly minimal contact with medical specialists. He had previously given very little thought to his dysmorphic appearance and did not recall undergoing any further investigations. He is the third of six children. Of his siblings, two of his younger sisters also had a similar physical appearance and ocular abnormalities, and similarly had not previously had any known respiratory disease.
Further information derived from the patient's family indicated that a presumptive diagnosis of ATD had been made during infancy based on the characteristic physical features. There was no reported respiratory failure in the neonatal period. No specific genetic testing had been performed to confirm this and the patient himself had not been made aware of this diagnosis. The patient's parents were known to be consanguineous and believed to be first or second cousins. Beyond the patient and two of his sisters, no other family members were known to be affected (Figure 3). On inpatient review by the clinical genetics team, the patient's physical characteristics were thought to be in keeping with this diagnosis, and a blood sample was taken should the patient want to confirm this with genetic testing in the future.

Family pedigree for the affected patient
The patient underwent a full range of cardiopulmonary investigations during his admission. The overall impression was of severe restrictive respiratory disease as a result of underlying congenital chondrodysplasia (forced expiratory volume in 1 s: 0.54 L [26% predicted], forced vital capacity: 0.62 L [23% predicted], forced expiratory ratio: 86%, total lung capacity: 1.82 L [43% predicted], residual volume: 1.14 L [61% predicted]). Diffusing capacity could not be performed due to insufficient lung volume. Resulting acute-on-chronic hypercapnic respiratory failure ensued (early morning arterial blood gas: pH 7.27, partial pressure of carbon dioxide [pCO2] 71 mmHg, partial pressure of oxygen [pO2] 46 mmHg, bicarbonate [HCO3] 32 mmol/L), complicated by pulmonary hypertension (estimated pulmonary artery pressure 47 mmHg, with mild right ventricular systolic impairment and preserved left ventricular function on transthoracic echocardiogram). Overnight unattended ward polysomnography showed no significant obstructive sleep apnoea, but there was nocturnal oxygen desaturation to a nadir of 87% presumably related to sleep hypoventilation in the clinical context albeit not confirmed with transcutaneous carbon dioxide monitoring. The 6-min walk test demonstrated oxygen desaturation on mobilization to 80% on room air. Muscle strength testing was within normal limits. The patient was commenced on nocturnal bilevel positive airway pressure (BPAP) with optimization of treatment settings planned in outpatient reviews, as well as exertional supplemental oxygen therapy. With ongoing BPAP therapy in the outpatient setting, there was normalization of pH and marked reduction of pCO2. Abdominal ultrasound demonstrated multicystic changes in the kidneys, but with preserved renal function. There was no other abdominal organ involvement detected.
DISCUSSIONFirst described in 1955, ATD is a recessively inherited chondrodysplasia, characterized by skeletal deformities and variable extra-skeletal organ involvement.1 With an incidence of 1 in 100,000–150,000 live births, ATD is a rare condition typified by a broad phenotypic spectrum of disease. The vast majority of cases are identified in infancy, proving to be fatal in a significant proportion.
The disorder is genetically heterogenous, and to date mutations in multiple different genes have been implicated.2 However, despite this heterogeneity, each of these genes have been found to similarly encode proteins necessary for ciliary intraflagellar support, a process that is critical for ciliogenesis and cell signalling.3 Cilia are essential structures found in various organs, including bone, liver, retina and kidneys. Disorders of cilia, or ciliopathies, are becoming increasingly implicated in causing human disease through disruption of cell signalling critical for normal human organ development.4 Two such genes that have recently been identified are the intraflagellar transport 80 (IFT80) and the intraflagellar transport (IFT) dynein-2 motor subunit (DYNC2H1) genes.2, 5 Mutations in the DYNC2H1 gene have been associated with fewer extra-skeletal manifestations, as was the case for our patient.2 However, severity of thoracic skeletal disease varies considerably even between siblings with identical DYNC2H1 mutations, highlighting the significant variability in disease phenotype.2
Skeletal deformities are the most common manifestations of ATD, and are typically the initial clinical and radiological abnormalities that prompt diagnosis. Prenatal diagnosis by ultrasound is possible, but difficult.6 Characteristic radiological features include a long, narrow, ‘bell-shaped’ thorax and shortened ribs, as demonstrated in Figure 2.7 It is this abnormal chest configuration that ultimately causes progressive lung restriction, respiratory failure and often premature death. Clavicles, pelvic and long bones are also commonly affected, with widening and shortening of limbs, and resulting short stature. Approximately 20% of patients have polydactyly of both hands and/or feet.8 Extra-skeletal manifestations are more variable and unpredictable, and can arise later in life. Renal, hepatic, pancreatic and retinal abnormalities can occur, with varying degrees of severity. Renal disease, typically cystic in nature, is thought to occur in approximately 30% of affected patients, and nearly 40% of these will develop end-stage renal failure.9 Our patient was known to have retinal involvement, and was found to have polycystic changes in his kidneys on abdominal ultrasound; however, renal function was preserved and no other abdominal organ involvement was detected.
The prognosis of ATD has historically been dire, with an estimated 60%–80% of affected patients dying in infancy or early childhood, typically as a result of respiratory failure.8 However, one more recent case series described 13 patients with known ATD, aged between 9 months and 22 years.8 Most of these patients had experienced respiratory problems in early childhood, and three died within the first 2 years of life. Extra-skeletal manifestations were again highly variable. However, this case series suggested that perhaps the prognosis is no longer as dire as previously reported, and indicated that respiratory disease often becomes less severe as the patient ages.8 Earlier disease detection, as well as improvements in more aggressive respiratory support in the antenatal period, are likely to have contributed to this improvement in mortality. Surgical intervention to manage respiratory restriction is also emerging as a potential therapeutic option for those children with severe thoracic wall disease.10 For those who do survive beyond childhood, close monitoring of lung function as well as regular screening for associated extra-skeletal manifestations are recommended.8 Surgical management for adults with ATD has been limited to date, although surgical reconstruction in adults with acquired forms of thoracic dystrophy has demonstrated symptomatic benefit.11 This may hold promise for those adults with congenital chondrodysplasias. Similarly, there is minimal literature to guide the use of BPAP in adults with ATD.
Adult identification of ATD is rare, with only one case report describing diagnosis in adulthood.12 Although ATD was suspected at birth in our patient, this particular case is novel given the patient and his two presumably affected sisters were previously unaware of the diagnosis, which is certainly remarkable. This unusual scenario also raises important ethical considerations regarding the concealment or obscuration of genetic disorders from the affected individuals, which has significant implications not only for the patient, but also for living family members and potential future offspring.
AUTHOR CONTRIBUTIONLachlan Stranks: primary author of the manuscript. Simone Barry: supervising clinician, editing of the manuscript. Aeneas Yeo: supervising clinician, editing of manuscript.
ETHICS STATEMENTThe authors declare that appropriate written informed consent was obtained for the publication of this manuscript and accompanying images.
留言 (0)