
記住我
Ectodermal dysplasias (EDs) are heterogenous genetic disorders with greater than 170 distinct syndromes currently described in the literature. Hallmark features of EDs include deficiency of mesoderm- and ectoderm-derived tissue including hair, skin, teeth and nails.1 Hypohidrotic EDs (HIEDs) are the most common EDs and are often characterized by sparse hair, abnormal dentition and hypohidrosis due to abnormal sweat glands.1, 2 Patients with these syndromes are at higher risk of respiratory disease compared to age- and gender-matched controls in the general population with mortality secondary to chronic and severe pulmonary infection.1 Here, we present the first familial cases of HIED receiving lung transplantation to highlight the sequelae of end-stage lung disease.
CASE REPORT Case 1Patient 1 was born at 38 weeks' gestation following an uneventful pregnancy and weighed 2100 g, secondary to intrauterine malnutrition. Early in childhood, he was noted to have marked dermatographia with lacy skin pigmentation along with a shrivelled appearance of several toenails and fingernails. Blocked tear ducts with recurrent bilateral epiphora and hypothyroidism manifested by age 6. Concurrently, the patient developed frequent suppurative chest infections, requiring numerous courses of antibiotics.3 Chest radiographs often showed mild to moderate degrees of lobar pneumonia with hyperinflation. A bronchial biopsy, performed at age 4, revealed microtubular derangement within the cilia of the respiratory epithelium. Skin biopsy showed an increased number of melanocytes and mast cells. Sweat gland counts per linear centimetre of the right-hand dermal ridge were markedly reduced. Given the constellation of symptoms and family history, the patient was diagnosed with an ED, specifically ANOTHER syndrome as previously described.3
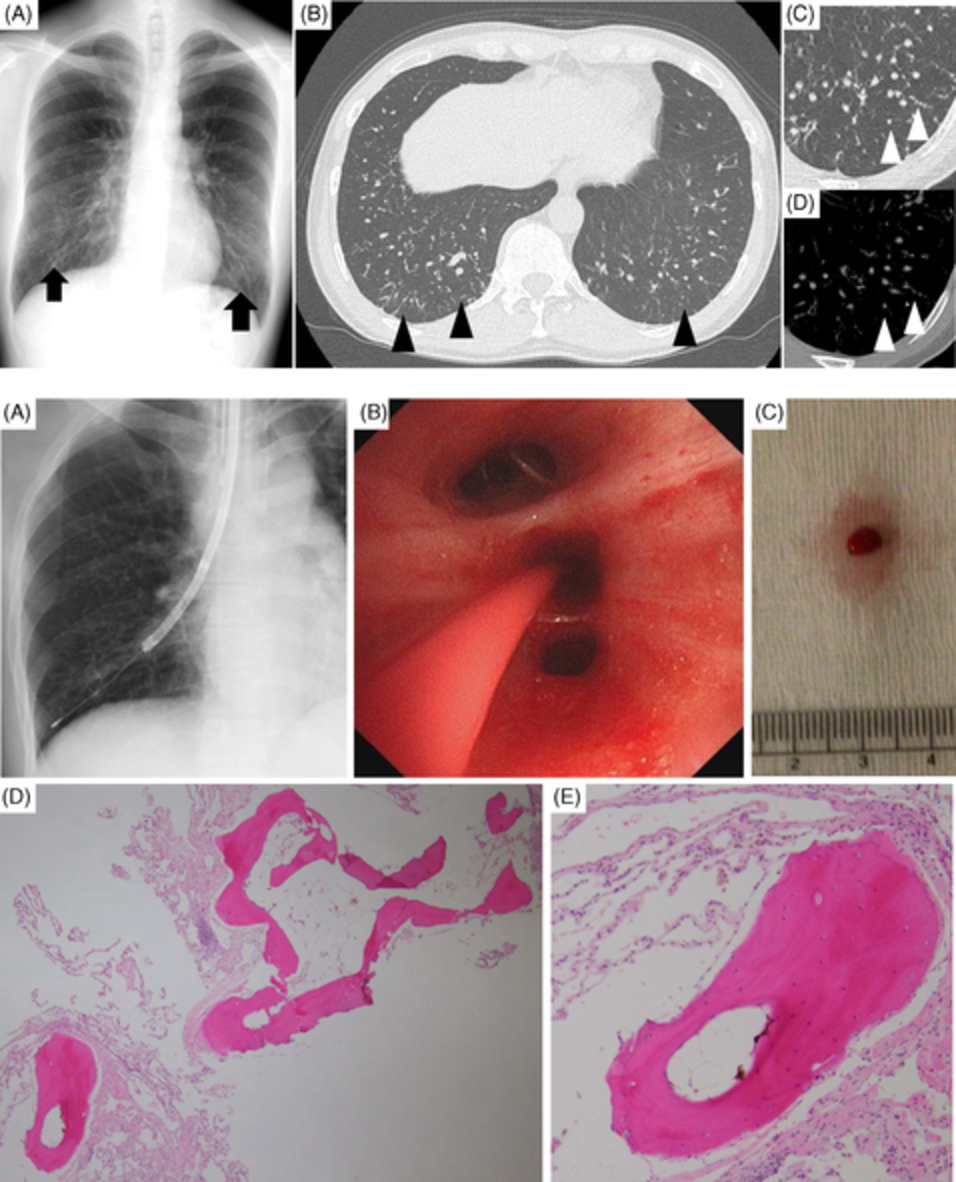
Patient 1 had a complicated course during early childhood leading to right lower and middle lobectomies at age 12 and removal of the remainder of the right lung by age 13 due to repeated infectious insults. He continued to develop chronic suppurative infections with further progression of respiratory disease throughout early adulthood. At age 32, the patient was evaluated and listed for lung transplantation based on progressive and irreversible decline in lung function (forced vital capacity [FVC] of 1.85 L [43%], forced expiratory volume in 1 second [FEV1] of 0.70 L [19%], FEV1/FVC of 38%, total lung capacity [TLC] at 4.23 L [82%], diffusing capacity for carbon monoxide [DLCO] of 9.7 ml/min/mmHg [30%]), need for supplemental oxygen (1–2 L/min at rest) and significant dyspnoea (Medical Research Council [MRC] of IV/V). The patient waited 206 days before a suitable lung became available. The patient underwent a left single lung transplant through median sternotomy with total cardiopulmonary bypass time of 141 min and donor ischaemic time of 125 min. Explant lung pathology demonstrated multifocal bronchiectasis with peribronchial fibrosis. Focal squamous metaplasia with reactive atypia and goblet cell hyperplasia were noted. Bronchiolectasis with obliterative bronchiolitis and centrilobular interstitial fibrosis were observed (Figure 1).

Composite explant lung pathological specimens of Patient 1. Patient 1 histology depicting inflamed airway bronchus (AW) with adjacent accompanying pulmonary artery (PA) (panel A; haematoxylin and eosin stain [H&E] at 20× magnification); inflamed dilated bronchiole (AW) with scarred and obliterated airway (arrows) adjacent to PA (panel B; H&E at 20× magnification); small dilated airway (AW) with scarring extending to abnormal thickened visceral pleura (VP) (panel C; H&E at 20× magnification); markedly inflamed small airways with intraluminal airway inflammation (panel D and insert E; H&E at 40× and 200× magnification, respectively)
Approximately 72 h after surgery, Patient 1 developed dense consolidation in the left lung in keeping with primary graft dysfunction or pulmonary oedema, empirically treated with diuresis and intravenous antimicrobial therapy. The patient then developed mild dehiscence at the left anastomosis, followed by bronchomalacia and stenosis requiring serial dilatations over the next several months. He additionally developed acute cellular rejection, determined by a decline in routine spirometry, treated with intravenous methylprednisolone and rabbit anti-thymocyte globulin.
Ten months after his lung transplant, the patient presented to hospital with massive pulmonary haemorrhage following bronchoscopy and was transferred to the intensive care unit. There, he had significant haematological derangement including anaemia, thrombocytopenia and schistocytes observed on peripheral smear with concomitant oliguric renal dysfunction. A diagnosis of thrombotic thrombocytopenic purpura was made with aggressive treatment with plasma exchange and discontinuation of the calcineurin inhibitor. Unfortunately, the patient developed progressive vasoplegia refractory to full medical intervention with development of septic shock and eventually succumbed to multi-organ failure.
Case 2Patient 2 was born 24 months earlier than his younger brother, Patient 1, at 37 weeks' gestation and weighed 2600 g. Patient 2 developed similar physical features to his brother with speckled skin pigmentation noted by 4 months of age with lack of scalp and eyebrow hair, hypohidrosis, dermatographia, ridged nails, epiphora and lack of dermal ridges in early childhood. Hypothyroidism was diagnosed at 2 years of age. Skin biopsy showed an increased number of melanocytes and mast cells. Given the family history with his younger brother, ANOTHER syndrome was again diagnosed. Their sister, 1 year younger than Patient 2, did not display any abnormalities.3 Despite the familial genetics, the clinical courses of the two brothers were vastly different as Patient 2 had relatively minor chest infections throughout adolescence and early adulthood.
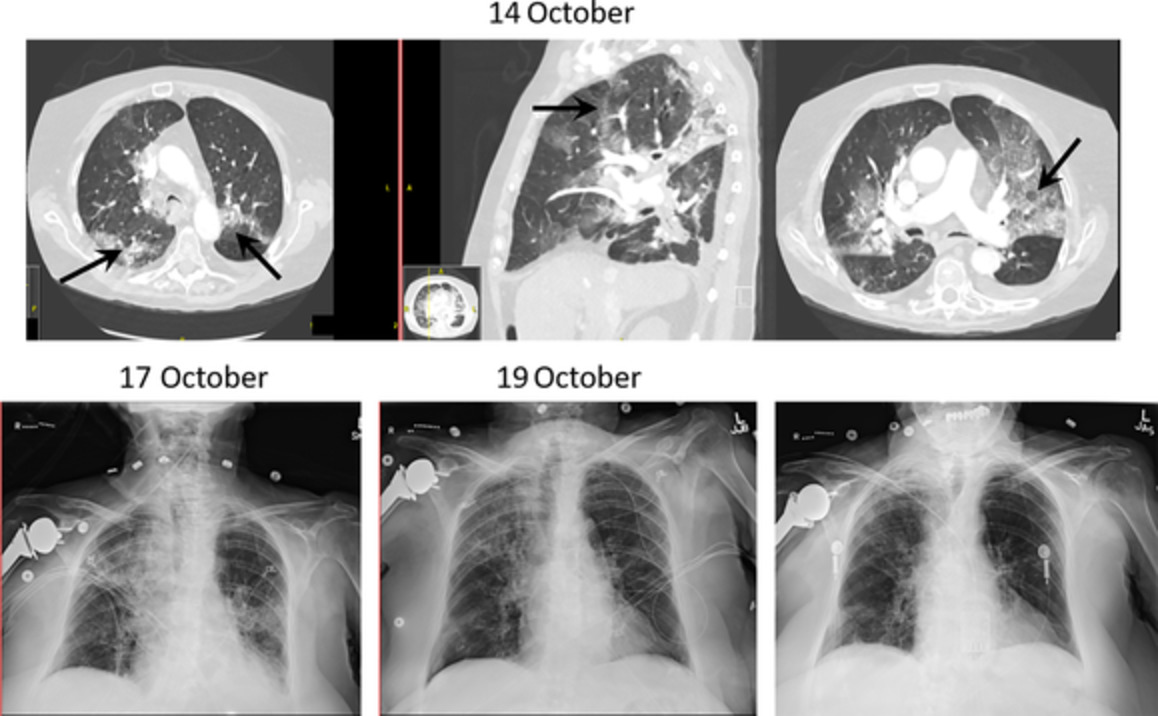
At age 46, Patient 2 presented to medical attention following a 2-year prodrome of increasing dyspnoea and cough. Subsequently, diffuse fibrotic changes in an atypical usual interstitial pneumonia (UIP) pattern were noted on his imaging (Figure 2). A connective tissue work-up was unremarkable, and he did not have any remarkable occupational or environmental exposures. His course during this time was complicated by spontaneous pneumomediastinum. Despite extensive investigations, antimicrobial treatments for bronchiectasis and chronic use of oral corticosteroids, his lung function deteriorated (FVC of 2.09 L [52%], FEV1 of 2.00 L [60%], FEV1/FVC of 96, TLC at 3.37 L [55%], DLCO of 12.55 ml/min/mmHg [57%]) with increasing need for supplemental oxygen (4 L/min at rest, 8–10 L/min with exertion). Due to his rapid irreversible progression, the patient was listed for lung transplantation. He subsequently underwent right and left lung transplantation through bilateral anterolateral (clamshell) thoracotomy. Explant lung pathology demonstrated diffuse bronchiectasis with background temporally heterogeneous active inflammatory fibrosing process with distribution consistent with UIP pattern (Figure 3). His first 4 years after transplantation have been uncomplicated.

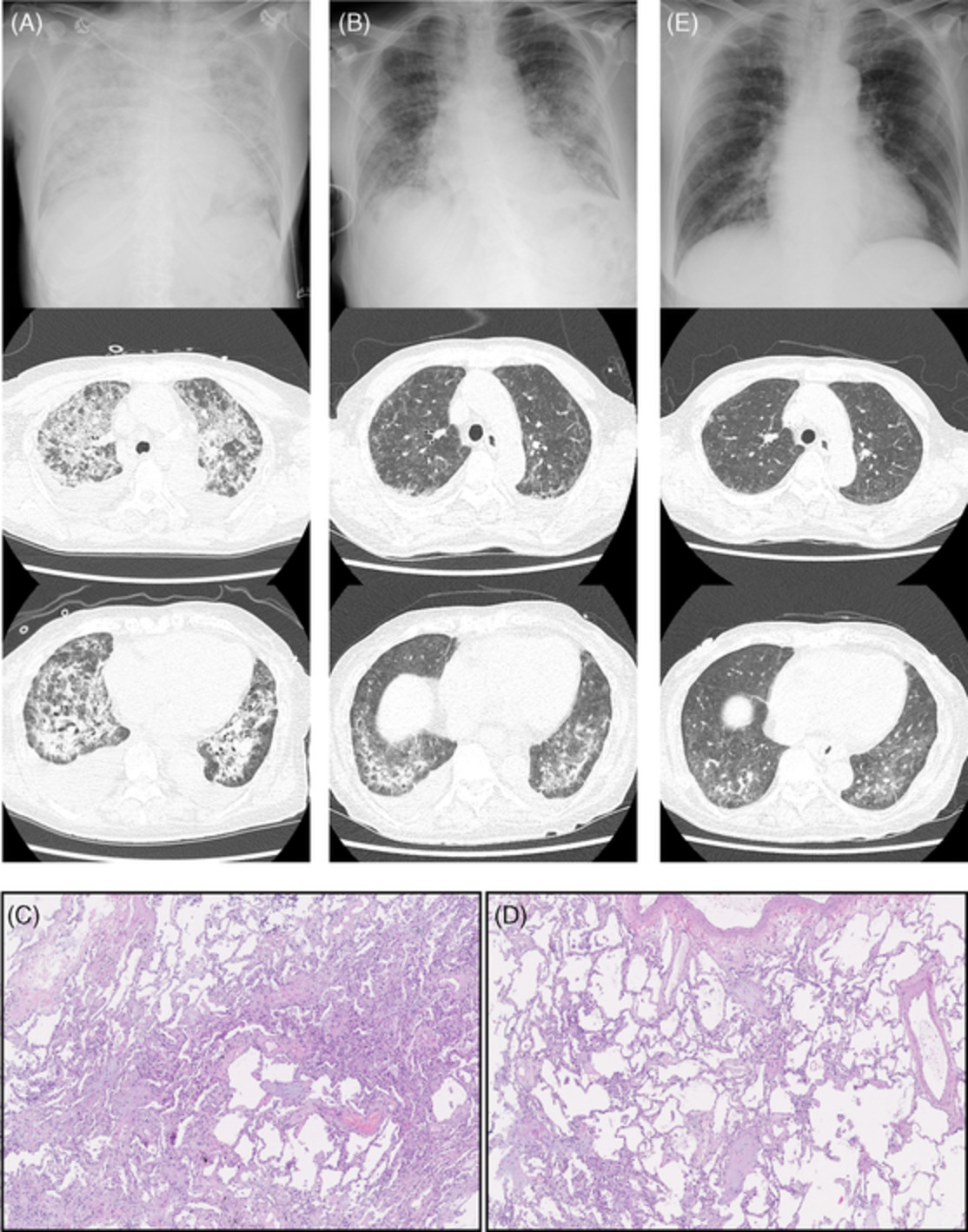
Progressive fibrotic lung disease. Axial computed tomography scan showing progression of Patient 2's lung disease from baseline (2007; A, B) to the time of initial presentation in May 2016 (C, D), October 2016 (E, F) and November 2016 prior to transplantation (G, H)

Composite explant lung pathological specimens of Patient 2. Patient 2 histology demonstrated inflamed dilated airway (AW) with adjacent pulmonary artery (PA) and peribronchial scarring (PBS) (panel A; H&E at 20× magnification); bronchiolectasis (AW) with less inflammation than Patient 1 but developing centrilobular scarring (arrows) (panel B; H&E at 20× magnification); further extensive scarring progressing into obliterative bronchiolitis (arrows) in smaller airways (panel C; H&E at 20× magnification); end-stage scarring and remodelling of normal lung (panel D and insert E; H&E at 20× and 200× magnification, respectively)
DISCUSSIONEDs are rare with incidence estimated at 1/100,000 births. While there are several entities within this classification, the most common is HIED, characterized by an absence or significant reduction of exocrine sweat glands. HIED is most common in males with an X-linked inheritance pattern.4 The genetic inheritance of HIEDs depends on the underlying genetic anomaly with X-linked inheritance associated with the ectodysplasin A (EDA, locus Xq12-q13.1, encoding ligand EDA-A1) gene and autosomal inheritance with EDA receptor (EDAR) and EDAR-associated death domain protein (EDARADD).5 Together, these genes are responsible for EDA production, a critical signalling protein in embryogenesis that discerns the ectoderm and mesoderm.6-13 The ectoderm–mesoderm interactions form the basis for development of skin, hair, nails, teeth and sweat glands. While HIED comprises several distinct features, pulmonary complications are frequent. The initial largest familial cohort to date of affected HIED individuals included 56 families in the UK and found that 65% reported lifetime incidence of asthma, 44% with recurrent respiratory infections in childhood and 26% with specific allergies.2 Mortality of males in the first year was reported at 21% and a total of 30% succumbed within the first 2 years, secondary to recurrent pulmonary infections.2 A follow-up study in Europe two decades later assessing 100 HIED children showed dramatic improvement in mortality to 2.1% in the first year of life, attributed to differing methodology and improved recognition in infancy amongst the medical community. Recurrent respiratory infections were reported in 73.5% of X-linked HIED.14
The presentation of respiratory manifestations in HIED is diverse. Anatomical defects, including the absence of mucous glands in the nasopharynx, trachea and respiratory tree, have been described on paediatric autopsies of several patients.1 Mucosal bronchoscopic sampling in adults showed similar pathology at the bronchus level. Otolaryngic manifestations, including atopic rhinitis, cleft palate and otitis media, have also been highlighted. Atopy was noted in up to 71% of HIED patients with significantly higher prevalence of asthma, allergies rhinoconjunctivitis and eczema compared to the general population controls (p < 0.001).15 From a mortality perspective, immunodeficiency and chronic respiratory recurrent infections are major contributors to recurrent inflammation, subsequent development of bronchiectasis and progressive respiratory failure.1 Compared to the general population, the prevalence of immunodeficiency is significantly higher in ED patients (6% vs. 0.05%; p < 0.01).1, 15 A recent 5-year follow-up study found the presence of obstructive airways disease, even in younger HIED children (11–35 months of age), highlighting the early manifestations of disease from infancy.16 Nitric oxide, produced by respiratory epithelial cells and macrophages, was a marker of inflammation in the lower respiratory tract with significantly higher fractional exhaled nitric oxide (FeNO) levels in both adults (p < 0.01) and children (p = 0.02) with HIED compared to healthy controls.17
Amongst HIEDs, ANOTHER syndrome is distinct with clinical features including Alopecia, Nail dystrophy, Ophthalmic complications, Thyroid disease, Hypohidrosis, Ephelides, Enteropathy and recurrent Respiratory tract infections. To our knowledge, this is the first familial case of lung transplantation for EDs. The first, and thus far, only case in the literature was described by Smythe et al. in 200018; the authors reported a 16-year-old patient with HIED with recurrent lower respiratory tract infections ultimately leading to respiratory failure. Bilateral lung transplantation was performed with an uncomplicated immediate post-operative period. Unfortunately, the patient developed Pseudomonas mastoiditis with septic shock 10 months post-transplant and died.18 While ANOTHER syndrome has been described with respiratory failure and requirement for lung transplantation, it is further important for the clinicians to be cognizant of the multi-system nature of this disease in the post-transplant setting. Alopecia and fair skin may predispose patients to skin malignancy in the setting of immunosuppression, stressing the need for regular dermatological evaluation.
While ANOTHER syndrome represents a diverse spectrum of clinical sequelae, despite the varied presentation of these two cases, the primary pathology in the explanted lung was similar. Patient 1 was explanted in the early third decade of life, over a decade earlier than Patient 2 by the time of transplantation. The pulmonary fibrosis observed on pathology, while developing, was limited in contrast to Patient 2 and may be explained by further evolution in the pathological spectrum. Interestingly, the fibrosis had features of UIP but with greater airway involvement than typical for idiopathic UIP. Thus, both pathological diagnoses demonstrate secondary UIP to bronchiectasis and repeated infections.
Transplantation in EDs is not limited to lungs. The first cardiac transplant case in EDs was described by Di Gesaro et al. in 2015.19 An 11-year-old paediatric patient with dilated cardiomyopathy and subsequent heart failure requiring inotropic support underwent a successful heart transplant. The post-operative course was complicated by a brief period of veno-venous extracorporeal membrane oxygenation for haemodynamic support. Long-term outcomes after 1 year were reported as successful.
Currently, there are no approved therapies for HIED with primary management directed towards the sequelae of the underlying disease. Recently, Fc-EDA, a recombinant protein composed of the receptor-binding portion of EDA1 and the fragment crystallizable (Fc) portion of immunoglobulin G1 (Fc-EDA), was developed with promising efficacy in animal models.20 Intravenous injection of Fc-EDA to newborn dogs with X-linked HIED restores growth of teeth, skin structures and mucous glands.21 Furthermore, intra-amniotic injections of Fc-EDA to pregnant mice partially restored the phenotype of the X-linked HIED newborn mice.22 Phase I (ClinicalTrials.gov Identifier: NCT01564225) and II (ClinicalTrials.gov Identifier: NCT01775462) studies completed in adults and neonates with Fc-EDA showed promising safety profiles but no statistically significant outcomes compared to placebo; they were limited by small sample size with a need for larger clinical trials to fully discern meaningful clinical outcomes.23
In summary, we present the first familial cases of lung transplantation in two brothers with end-stage lung disease from HIED. While a rare disorder, EDs may be underdiagnosed due to the heterogeneity of disease presentation. These cases highlight the high acumen required by the clinicians and propensity for significant clinical complications. Pulmonary disease in HIED portends a poor prognosis, and lung transplantation may be considered in these patients with the most severe pulmonary involvement. Patient 2 represents the longest surviving post lung transplant recipient of the three worldwide cases reported in the literature. Furthermore, these cases stress the importance of comprehensive genetic and/or congenital evaluation in individuals with end-stage lung disease. In those patients with multiple respiratory infections, aggressive early medical intervention is critical as ongoing infectious cycles may lead to bronchiectasis and fulminant pulmonary fibrosis and scarring.
ACKNOWLEDGMENTSWe thank the patients and their family for granting permission to share their stories.
AUTHOR CONTRIBUTIONChristina S. Thornton, Lakshmi Puttagunta, Douglas Helmersen, Mitesh V. Thakrar, Jayan Nagendran and Rhea A. Varughese were all directly involved in care of one of the patients. Dale Lien was directly involved in the care of both patients. Christina S. Thornton and Rhea A. Varughese contributed to data acquisition by chart review. Lakshmi Puttagunta provided pathological specimen pictures and interpretation. Christina S. Thornton, Lakshmi Puttagunta and Rhea A. Varughese drafted the manuscript. All authors made substantial contributions to the conception and design of this work; contributed to the interpretation of the data; and critically revised it for important intellectual content, approved the version to be published and agreed to be accountable for all aspects of the work.
ETHICS STATEMENTThe authors declare that appropriate written informed consent was obtained for the publication of this manuscript and accompanying images.
留言 (0)