
記住我
Interstitial pneumonia with autoimmune features (IPAF) is a term proposed in 2015 to describe idiopathic interstitial pneumonias that have clinical features of connective tissue disease (CTD), but do not meet the established criteria for CTD.1 One of the morphological domains in the criteria for IPAF include the pathological pattern observed in surgical lung biopsy (non-specific interstitial pneumonia [NSIP], organizing pneumonia [OP], NSIP with OP overlap, lymphocytic interstitial pneumonia, interstitial lymphoid aggregates with germinal centres or diffuse lymphoplasmacytic infiltration). Li et al. suggested that IPAF should receive early immunosuppressive treatment.2 However, how and when to treat IPAF has not been determined. Here, we report a spontaneous improvement case of IPAF.
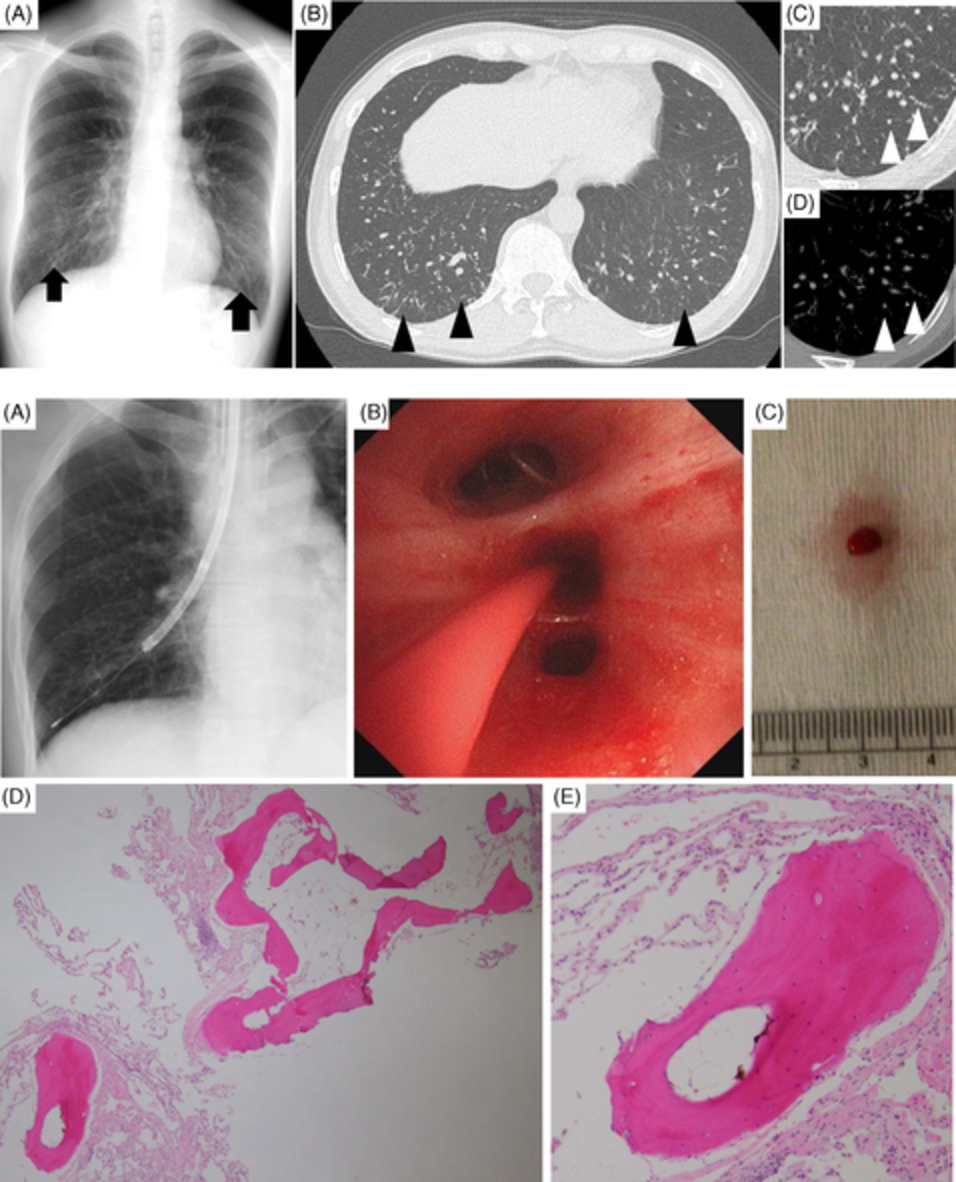
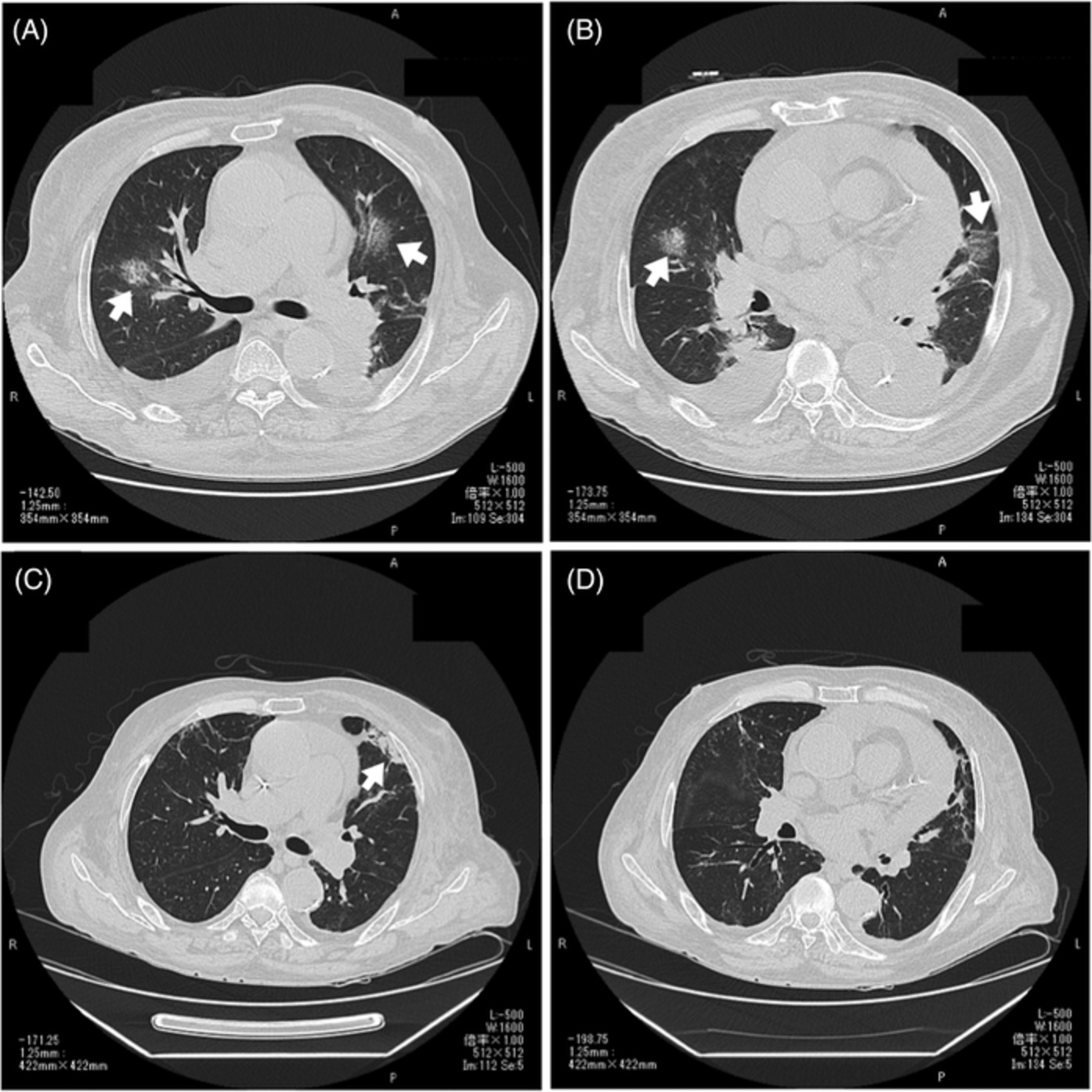
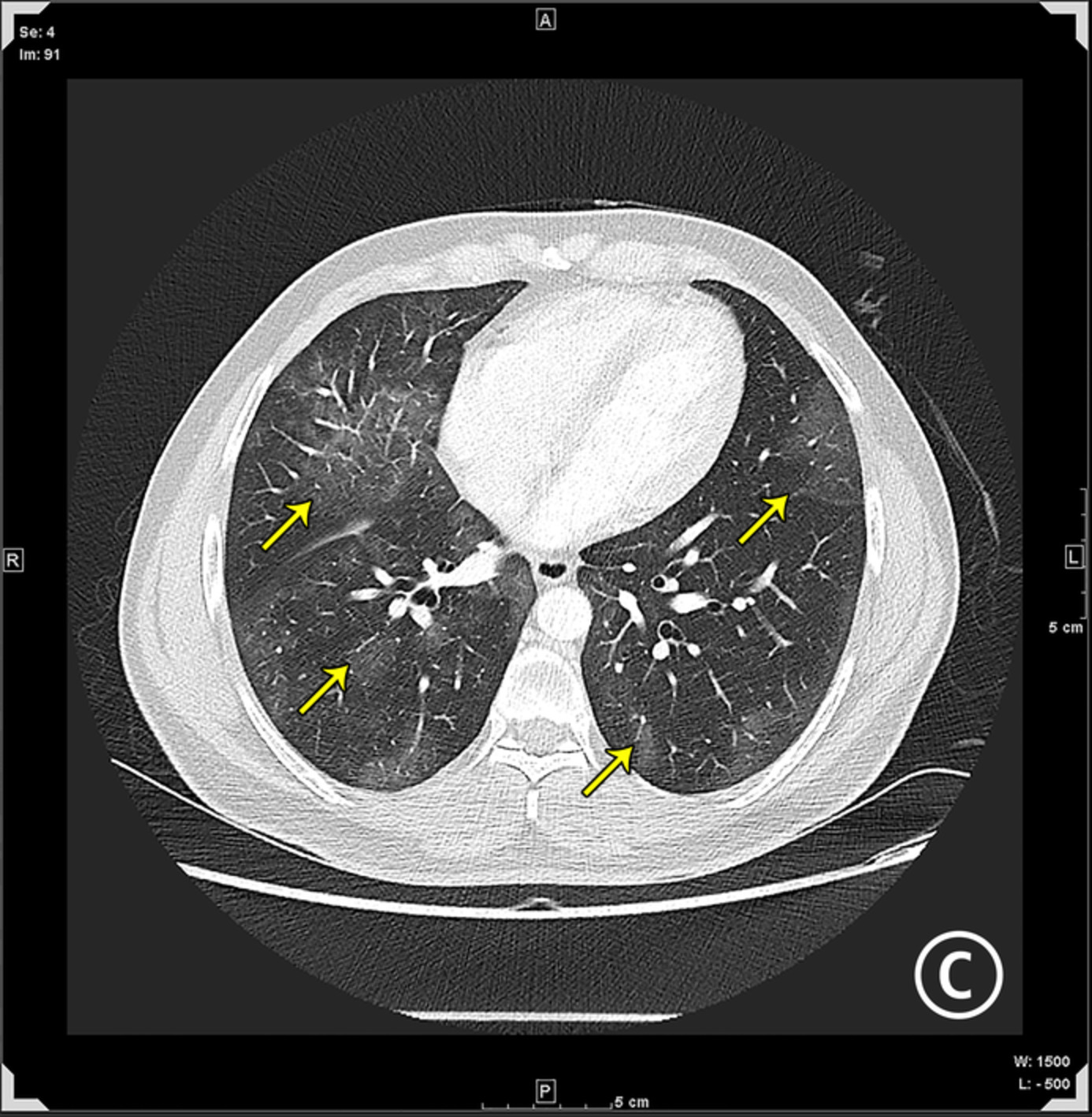
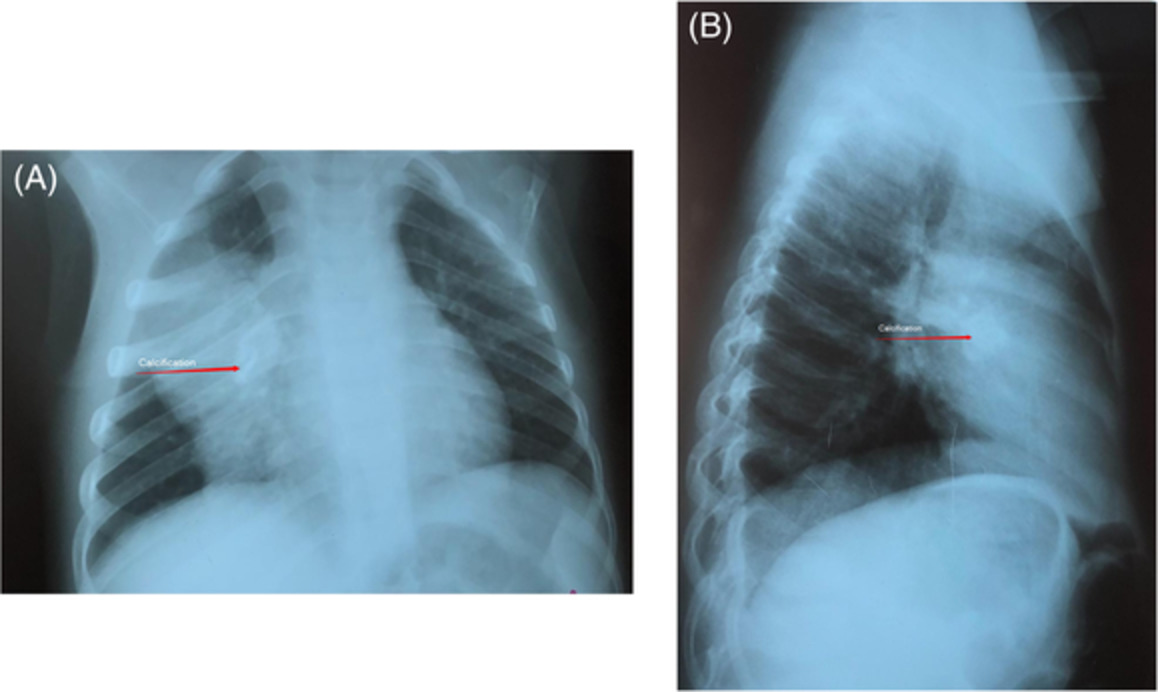
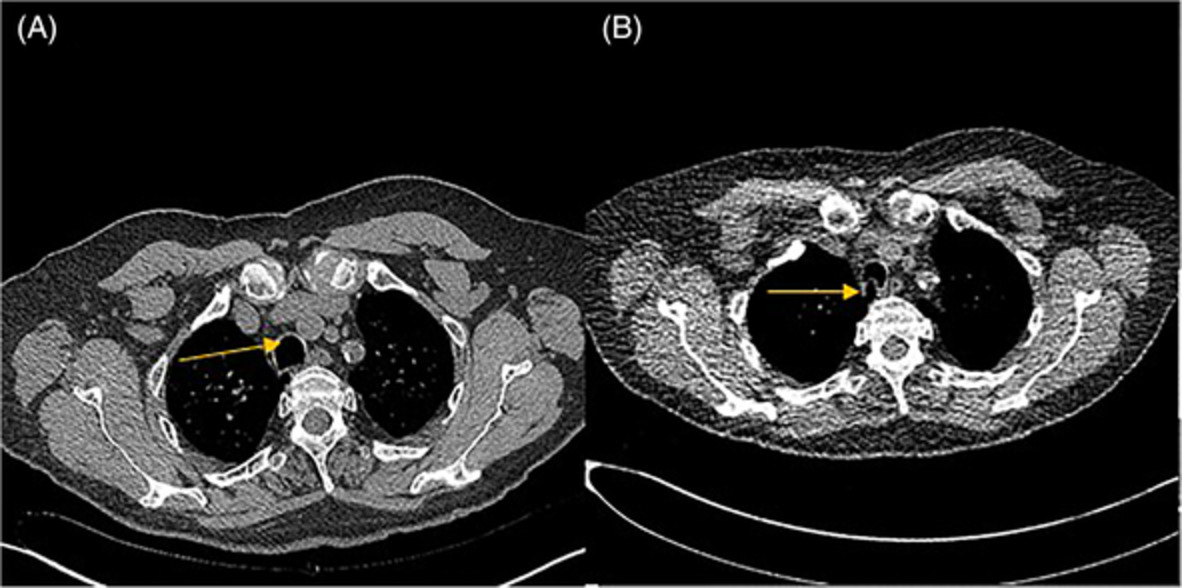
CASE REPORTA 58-year-old man developed dry cough and dyspnoea prior to presentations at our hospital. Three weeks before, he had developed symptoms of elevated fever and visited a local clinic, but the rapid flu test yielded negative results. He visited another hospital due to cough and progressive dyspnoea, and high-resolution computed tomography (HRCT) of the chest showed reticular regions and ground-glass opacities, predominantly in bilateral lower lung lobes (Figure 1A). He was then referred to our hospital. He had no history of exposure to asbestos and had previously smoked 1.25 packs of cigarettes a day for 37 years. He had no history of bird rearing or drug allergies and no family history of interstitial lung disease. Physical examination revealed periungual erythema, periungual epithelial extensions and nail fold bleeding, but no sclerosis. Fine crackles were audible on his back. Resting and ambulatory percutaneous oxygen saturation percentages in room air were 94% and 82%, respectively. Laboratory data showed leucocytosis (8100/μl) with neutrophilia (71.2%). C-reactive protein was 0.75 mg/dl. Serum lactate dehydrogenase concentration was elevated to 328 IU/L (normal range, 120–220 IU/L) and serum Krebs von den Lungen (KL)-6 level was elevated to 1977 U/ml (normal range, <500 U/ml). The anti-nuclear antibody (1:640 titre, discrete speckled) and the anti-centromere antibody were positive. Pulmonary function testing results showed a forced vital capacity (FVC) of 2.72 L (69.4% of predicted value) and diffusing capacity of the lung for carbon monoxide was 8.62 ml/min/torr (40.5%). The patient met neither the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) criteria for systemic scleroderma,3 nor the very early diagnosis of systemic sclerosis (VEDOSS) criteria.4 We performed transbronchial cryobiopsy from the left lower lung lobe using flexible bronchoscope under deep sedation, revealing a cavity in the left lower lung lobe on HRCT after the procedure (Figure 1B). A predominance of lymphoid cells was confirmed in bronchoalveolar lavage fluid (BALF) from the inferior lingular segment; lymphocytes, 50%; neutrophils, 1%; eosinophils 8%; macrophage, 41%; and CD4/CD8 ratio, 0.20. No microorganisms were cultured in BALF. A cryobiopsy specimen obtained from the left lower lung lobe revealed mild thickening of the alveolar wall without vasculitis (Figure 2). The histology was compatible with cellular NSIP. Symptoms and ground-glass opacities slightly improved, and the patient declined immunosuppressive treatment. Repeated HRCT after 6 months showed an apparent reduction in the area of ground-glass opacities (Figure 1C). The FVC improved from 2.72 to 3.47 L, representing a 16.2% improvement. Serum KL-6 decreased from 1977 to 531 U/ml, and cough and dyspnoea disappeared. Furthermore, periungual erythema and nail fold bleeding improved.

High-resolution computed tomography (HRCT) images and histological findings. (A) HRCT image at first visit. (B) HRCT image after transbronchial cryobiopsy. Arrowhead shows a cavity by cryobiopsy. (C) HRCT image after 6 months

Histological findings for transbronchial cryobiopsy specimen of the left lower lobe. Images show moderate thickening of the alveolar walls (haematoxylin and eosin staining). (A) Low magnification, (B) High magnification
DISCUSSIONThis patient met the IPAF criteria,1 and pathological analysis of cryobiopsy specimen demonstrated cellular NSIP. The diagnostic criteria for systemic sclerosis were revised in 2013 by the ACR/EULAR.3 The present patient met neither the ACR/EULAR criteria nor the VEDOSS criteria,4 but showed positive results for the anti-centromere antibody, presence of periungual erythema, periungual epithelial extension and nail fold bleeding.
In the present case, FVC and KL-6 improved spontaneously without administration of corticosteroids and immunosuppressants, while transbronchial cryobiopsy led to the diagnosis of cellular NSIP. The natural courses of cellular NSIP that meet the criteria for IPAF are not yet fully understood. To the best of our knowledge, this is the first report that cellular NSIP spontaneously improved that meet the criteria for IPAF with positive result for the anti-centromere antibody. It is important for the physician to consider the appropriate indications for the administration of corticosteroids and immunosuppressants in patients who meet the IPAF criteria.
Periungual erythema and nail fold bleeding sometimes improve, but usually do not improve in patients with systemic sclerosis. In the present case, these finger abnormalities and interstitial lung disease improved simultaneously. It is unknown how often IPAF progress to CTD, and what differences are present in the pathogenesis between IPAF and CTD. The temporal deteriorations of symptoms and signs might be caused by aberrant immune reactions for viral infection or environmental triggers. However, further research is needed to confirm such hypothesis.
The present case is categorized as idiopathic NSIP according to the current guidelines and improved spontaneously. IPAF remains a research diagnosis with little consensus on optimal treatment strategies. Accumulation of more cases is necessary for understanding and treating IPAF.
CONFLICT OF INTERESTNone declared.
AUTHOR CONTRIBUTIONHirotsugu Ohkubo and Akiko Nakano equally contributed to the study. They drafted the submitted article and take responsibility for the integrity of the data and the accuracy of the data analysis. Kohei Fujita, Yoshiyuki Ozawa and Akio Niimi contributed to the interpretation of the manuscript. Takayuki Murase contributed as a pathologist.
ETHICS STATEMENTThe authors declare that appropriate written informed consent was obtained for the publication of this case report and accompanying images.
REFERENCES
1Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015; 46: 976– 87. 2Li Y, Zheng Z, Han Q, Li Z, Xie R, Zhang R, et al. IPAF should receive early treatment for sharing similar clinical characteristics as CTD-ILD: a report from 273 Chinese patients. Clin Rheumatol. 2020; 39: 3817– 23. https://doi.org/10.1007/s10067-020-05149-6 3van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2013; 65: 2737– 47. 4Matucci-Cerinic M, Allanore Y, Czirják L, Tyndall A, Müller-Ladner U, Denton C, et al. The challenge of early systemic sclerosis for the EULAR Scleroderma Trial and Research group (EUSTAR) community. It is time to cut the Gordian knot and develop a prevention or rescue strategy. Ann Rheum Dis. 2009; 68: 1377– 80.
留言 (0)