
記住我
The epithelial barrier is essential for maintaining tissue homeostasis and protecting against exogenous insults. Loss of barrier function results in severance of the intricate structural framework of the epithelia and increased susceptibility to noxious stimuli such as bacterial infection and inflammation (Groeger and Meyle, 2015; Rogers et al., 2023). Bacterial pathogens are known to exploit transcytosis, as well as other uptake mechanisms like internalization or paracytosis (intercellular passage), to penetrate epithelial and other tissue barriers. These strategies enable them to reach underlying niches or access the intra- and sub-epithelial spaces, facilitating their spread (Kaper et al., 2004; Edwards and Massey, 2011; Nikitas et al., 2011; Zhu et al., 2024). To this end, it is essential to elucidate cellular phenomena and understand the key regulatory events that govern the integrity of a well-controlled epithelial barrier. This may pave the way for new therapeutic avenues that will enable the development of targeted interventions to mitigate barrier impairment and, ultimately, restore its functionality in pathophysiological conditions.
The epithelial barrier acts as a physical and immunological barrier, separating the internal milieu from the external environment. It represents a composite network of cell adhesion molecules (CAMs), such as adherens junctions (AJs), tight junctions (TJs), and desmosomes, which collectively maintain the epithelial polarity and barrier microarchitecture (Figure 1) (Adil et al., 2021). Infectious agents dissociate these junctional complexes and destabilize the selective permeability and structural coherence, facilitating barrier breach and pathogen invasion into the interstitial tissues (Groeger and Meyle, 2015; Rogers et al., 2023).
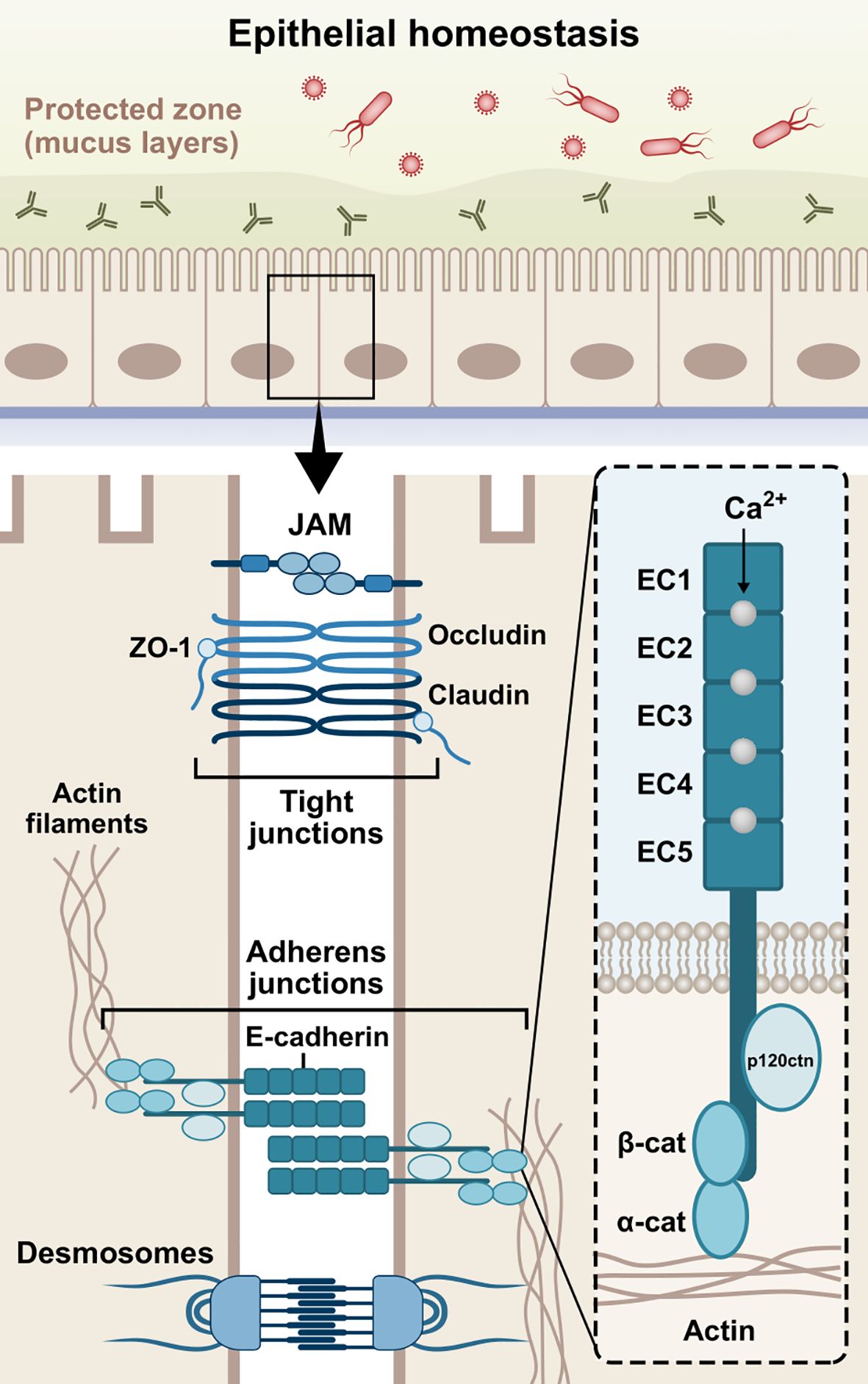
Figure 1. Schematic structure of E-cadherin. The extracellular domain contains five 110 amino acids repeated regions (EC1-EC5), in which the Ca2+ ions work as inter-domain linkers to stabilize the adhesive interactions between adjoining cells. The single-pass transmembrane region of E-cadherin transverses the phospholipid bilayer and facilitates the interactions of the extracellular domains with the cytoplasmic domain. The cytoplasmic tail consists of roughly 150 amino acids and regulates downstream signaling pathways. Cadherins initially form cis-dimers on the same cells, followed by the formation of trans-dimers with cadherins on adjacent cells, establishing adhesion across the paracellular space. The three domains are involved in the epithelial barrier function via formation and stabilization of AJs. JAM, junctional adhesion molecule; ZO-1, zonula occludens-1; β-cat, β-catenin; α-cat, α-catenin; p120ctn, p120 catenin; EC1-5, extracellular cadherin repeats 1-5.
The formation of AJs requires the presence of Ca2+-dependent transmembrane adhesion glycoproteins, named cadherins. They act more than mere cell glue designated to serve mechanical cohesion between adjacent cells; they orchestrate junctional assembly and inter-junctional communication, and participate in signaling pathways that regulate cellular behavior, such as proliferation, migration, differentiation, epithelial repair, wound healing, or even morphogenesis (Stockinger et al., 2001; Gumbiner, 2005; Halbleib and Nelson, 2006; Van Roy and Berx, 2008, 2008; Van Den Bossche et al., 2012). The most well-studied cadherins are the classical vertebrate cadherins, which have been named based on the tissue in which they are expressed. Neuronal cells mainly express N-cadherin (CDH2), while epithelial cells highly express E-cadherin (CDH1) (Rajwar, 2015; László and Lele, 2022; Kadeh et al., 2023). P-cadherin (CDH3) has been found in breast tissue, skin, and hair follicles, as well as lungs and placenta among others (Vieira and Paredes, 2015). In addition, VE-cadherin (CDH5) is specifically expressed in vascular endothelial cells, where it controls their behavior during angiogenesis (Nan et al., 2023), while K-cadherin (CDH6) is primarily found in the kidney (Cho et al., 1998; Thedieck et al., 2005) and R-cadherin (CDH4) mainly in the brain (Martinez-Garay et al., 2016). Interestingly, it has been found that E-cadherin is also present in immune cells, such as dendritic cells (DCs), macrophages, and T-cells (Riedl et al., 2000; Van Den Bossche et al., 2012; Van Den Bossche and Van Ginderachter, 2013; Charnley et al., 2023; Davies et al., 2024). E-cadherin is a type-I cadherin encoded by the CDH1 gene on chromosome 16q22 (Van Roy and Berx, 2008). The E-cadherin molecule is composed of three distinct structural domains, namely an extracellular domain, consisting of 5 repeated regions (EC1-EC5), which engages in homotypic (cis- and trans-dimers) and heterotypic cell-cell interactions, a single-pass transmembrane domain, and a cytoplasmic tail which regulates downstream signaling (Gumbiner, 2005; Hulpiau and Van Roy, 2009). The domain structure of E-cadherin is illustrated in Figure 1.
This review aims to provide a comprehensive overview of E-cadherin as a major junctional molecule with respect to tissue homeostasis and its dysregulation in the etiopathogenesis of bacterial infections, inflammatory, and other conditions. Initially, we report the molecular underpinnings of E-cadherin-directed cell-cell adhesion and relevant signaling pathways in homeostasis. Next, we describe E-cadherin-mediated mechanisms in bacterial infections, inflammation, and other diseases by delving into alterations in E-cadherin expression, localization, and functionality. Furthermore, we highlight the clinical implications of epithelial barrier dysfunction and the mechanistic and immunological involvement of E-cadherin in disease across various tissues, emphasizing numerous infection examples and inflammation models. Lastly, we examine potential therapeutic strategies targeting junctional compounds and E-cadherin to enhance and restore epithelial barrier integrity and tackle infection.
2 Homeostatic regulation of the epithelial barrier via E-cadherinE-cadherin plays a vital role in tissue homeostasis by contributing to selective, semi-permeable barrier structure features via sealing the intercellular spaces between the cells and promoting the formation of AJs (Wheelock and Johnson, 2003; Takeichi, 2014). Herein, we aim to report E-cadherin-mediated mechanisms that are involved in the barrier assembly and are responsible for maintaining epithelial homeostasis (Figure 2).
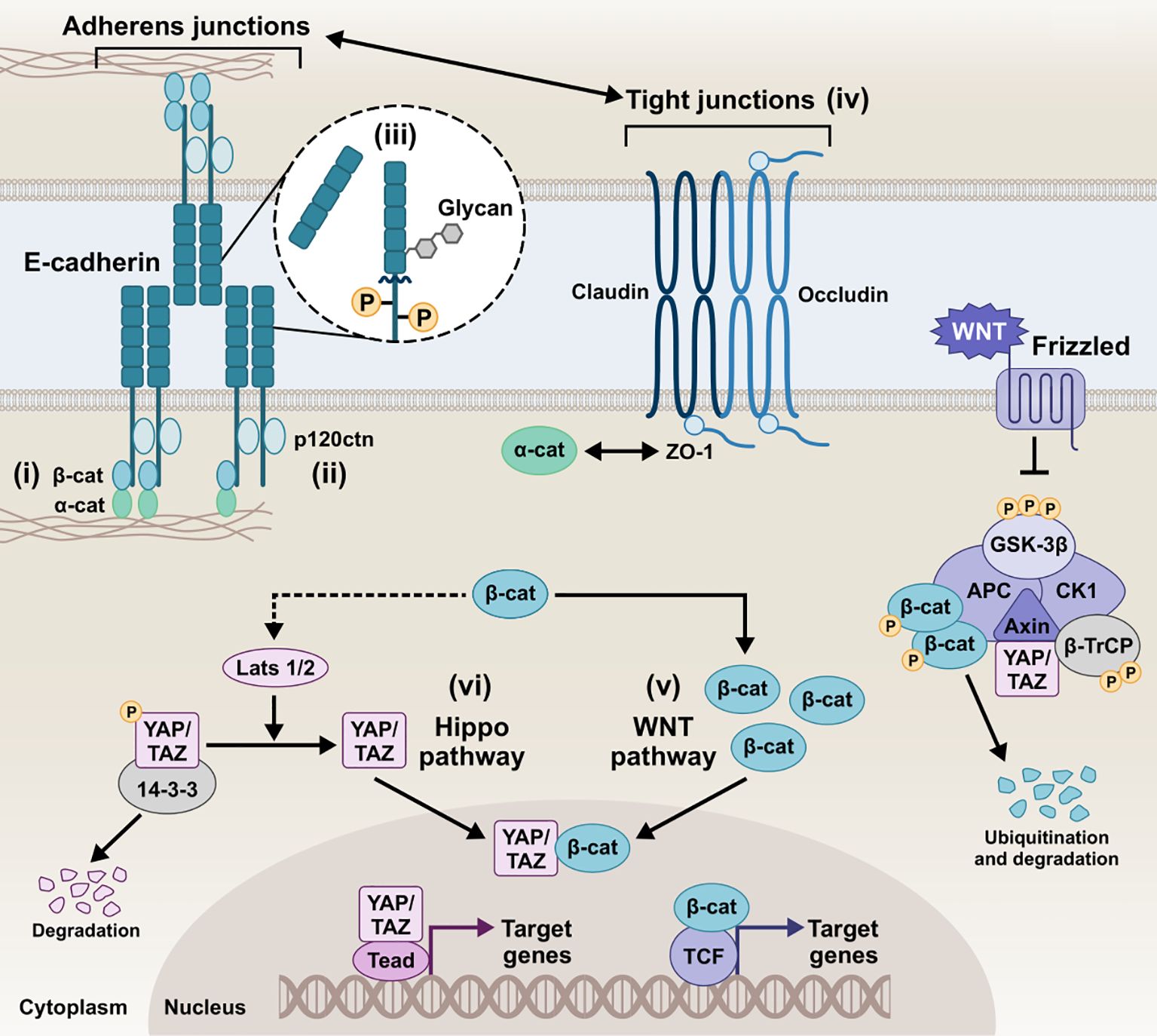
Figure 2. Schematic representation of the E-cadherin interactions and homeostatic mechanisms involved in the regulation of the epithelial barrier of epithelial barrier. i) E-cadherin/β-catenin/actin complex, ii) E-cadherin/p120ctn complex, iii) E-cadherin post-translational modifications, iv) Tight junctions and E-cadherin, v) E-cadherin and Wnt pathways, vi) E-cadherin and Hippo pathway, collectively play a critical role in tightly regulating cellular behavior and intercellular communication. Intricate modulation of the AJs integrity, downstream signaling, and overall epithelial barrier function preserves homeostatic conditions in the host tissue. β-cat, β-catenin; α-catenin, α-catenin; p120ctn, p120 catenin; ZO-1, zonula occludens 1; GSK-3β, glycogen synthase kinase-3 beta; CK1, casein kinase 1; APC, anaphase-promoting complex; β-TrCP, beta-transducin repeats-containing protein; YAP, yes-associated protein; TAZ, transcriptional co-activator with PDZ-binding motif; LATS1/2, large tumor suppressor kinase 1/2; Tead, transcriptional enhanced associate domain; TCF, T-cell factor.
2.1 E-cadherin/β-catenin/actin complexThe canonical pathways involved in AJ assembly demonstrated the E-cadherin clustering controlled by the intracellular tail and the coupled actin cytoskeleton (Yap et al., 1998; Wu et al., 2015; Biswas and Zaidel-Bar, 2017). Specifically, the C-terminus of the intracellular tail interacts with a group of adaptor proteins called armadillo catenins, namely β-catenin and plakoglobin (γ-catenin), which anchor E-cadherin to the peri-junctional actin cytoskeleton. γ-Catenin is primarily localized at desmosomes and AJs, interacting with desmogleins/desmocollins and cadherins, respectively, and can compensate for β-catenin loss at AJs without disrupting desmosomal integrity (Diane Wickline et al., 2013). The E-cadherin-catenin complex -known as CCC- is composed of β-catenin (or plakoglobin), which directly tethers via its central Armadillo domain to the cytosolic tail of E-cadherin and via the N-terminal domain to α-catenin, which in turn links the compound to the actin filaments (F-actin) (Pećina-Šlaus, 2003; Kobielak and Fuchs, 2004; Hulpiau and Van Roy, 2009). The binding of α-catenin to F-actin requires α-catenin homodimers, whereas α-catenin binds to E-cadherin/β-catenin complex in its monomeric form. EPLIN (i.e., epithelial protein lost in neoplasm) represents the missing link between the CCC and the apical circumferential actin belt, coupling cortical actin filament bundles to the monomeric α-catenin of the assembly (Abe and Takeichi, 2008).
2.2 E-cadherin/p120ctn complexA highly conserved sequence in the juxtamembrane domain of E-cadherin is responsible for coupling with another catenin, named p120 catenin (p120ctn), whose binding is fundamental for the AJ assembly (Thoreson et al., 2000; Van Roy and Berx, 2008). p120ctn acts as a master regulator of E-cadherin’s cell surface delivery and functional integrity by inhibiting internalization pathways that promote E-cadherin degradation and facilitating plasma membrane recycling (Davis et al., 2003). It has been reported that the juxtamembrane region primarily mediates the lateral clustering of cadherin molecules, further reinforcing the role of p120ctn as a key contributor to cluster formation and adhesion strengthening (Yap et al., 1998). Moreover, p120ctn is an important mediator for the Rho-associated protein kinase (ROCK)/E-cadherin interaction. ROCK is a serine-threonine kinase involved in the regulation of cadherin function. Constitutive activation of ROCK leads to disruption of AJs, whereas pharmacological inhibition of ROCK promotes AJ stability (Wójciak-Stothard et al., 2001; Grothaus et al., 2018).
2.3 Post-translational eventsPost-translational processing of E-cadherin, most prominently including phosphorylation, O-glycosylation, N-glycosylation, and proteolytic cleavage, has been extensively described to dictate its function and redistribution dynamics. Serine phosphorylation of the β-catenin-binding domain, for instance, has been reported to be constitutive to cadherin-catenin complex formation and stabilization by increasing β-catenin binding affinity and regulating E-cadherin’s biosynthesis and trafficking (McEwen et al., 2014). Effector phosphorylation of p120ctn and β-catenin also seem to -inversely- contribute to the E-cadherin/catenin association and partly control E-cadherin’s surface stability (Roura et al., 1999; Fukumoto et al., 2008). Cytoplasmic O-glycosylation (O-GlcNAc) of newly synthesized E-cadherin regulates its secretory path, causing retention in the endoplasmic reticulum and cell surface transit arrest. In its absence, unimpeded export to the membrane delays apoptosis and rescues E-cadherin recruitment to adhesion sites (Geng et al., 2012). Ectodomain N-glycosylation constitutes the most prevalent post-translational modification, boasting four potential sites (two in EC4 and two in EC5) in the extracellular domain of human E-cadherin. In addition to E-cadherin folding and trafficking, N-glycan remodeling can be instrumental to functional junction organization, with the extent of N-glycan branching/complexity negatively associating with adhesive strength (Pinho et al., 2011). Another functionally-impairing post-translational event E-cadherin can undergo is proteolytic truncation by endogenous proteases, which more prominently results in the release of soluble E-cadherin (sE-cad) fragments, as discussed in more detail below. sE-cad is approximately 80 kDa in size, generated by α-secretase cleavage on the extracellular face of the plasma membrane, which is catalyzed by various proteases, including matrix metalloproteinases (MMPs), members of a disintegrin and metalloproteinase (ADAMs) family, plasmin, and kallikrein 7 (David and Rajasekaran, 2012). The shed sE-cad fragment can diffuse into the extracellular environment, where it retains the ability to form homophilic bonds and pair with intact, full-length molecules, interfering with the function of adhesion-competent E-cadherin. Moreover, it can chemotactically anchor E-cadherin on migrating cells and upregulate MMPs, thereby further destabilizing epithelial integrity (Samuels et al., 2023). Ectodomain shedding disrupts the intact E-cadherin junctional complexes, with circulating sE-cad harboring biological effect amplification in the context of proliferative and survival/apoptotic resistance signals, migratory and invasive abilities due to loss of barrier function, inflammation, and tumorigenesis (Grabowska, 2012). The remaining membrane-bound C-terminal fragment of E-cadherin (38 kDa, E-cad/CTF1) can then be cleaved by a γ-secretase/presenilin-1/2, injecting a 33-kDa E-cad/CTF2 fragment into the cytosol. This unleashes β-catenin which can promote the oncogenic canonical Wnt pathway, with E-cadherin sheddase matrilysin (MMP-7) among the transcriptional targets. Also, p120ctn remains E-cadherin-bound and can mediate E-cad/CTF2 translocation to the nucleus and subsequent DNA binding, where E-cad/CTF2 modulates p120ctn-Kaiso-mediated pathway to suppress apoptosis (Ferber et al., 2008). In addition to fragmentation into CTF1 and CTF2, generation of a 29kDa E-cad/CTF3 by caspase-3 has been observed in apoptosis and cancer progression (Craig and Brady-Kalnay, 2011; Yang et al., 2017).
2.4 E-cadherin and tight junctions (TJs)Tungal et al. demonstrated that E-cadherin is crucial for maintaining epithelial barrier function in vivo by regulating TJ formation and stability. Specifically, E-cadherin coordinates the trafficking and positioning of TJ proteins, facilitating the localized integration of key molecules such as the cytoplasmic scaffolding zonula occludens 1 (ZO-1) and claudins, a family of integral membrane proteins that form TJs (Tunggal et al., 2005; Maiers et al., 2013). The communication between AJs, mediated by E-cadherin, and TJs plays a vital role in establishing inter-junctional co-dependence and directing the initial architecture of the epithelial barrier (Ando-Akatsuka et al., 1999; Lázaro et al., 2002; Tunggal et al., 2005).
The functional coupling of AJs and TJs is essential for the maturation of AJs and the early development of TJs. Early studies found that ZO-1 mobilization to the plasma membrane is mediated by catenins, enabling ZO-1 to co-distribute in areas segregated by E-cadherin (Rajasekaran et al., 1996). ZO-1, a key marker of TJs, is closely associated with AJs and the cadherin-catenin complex, transiently binding with α-catenin in nascent junctions (Maiers et al., 2013; Campbell et al., 2017). Knockdown of E-cadherin using siRNA has been shown to reduce ZO-1 expression and lower epithelial resistance in bronchial epithelial cells (Heijink et al., 2010). Additionally, loss of E-cadherin disrupts the organization of ZO-1 and F-actin, as E-cadherin-dependent mechanical circuits play a role in integrating force transduction and signaling pathways that drive junctional polarization necessary for functional epithelial barrier formation (Rübsam et al., 2017).
E-cadherin also regulates epidermal growth factor receptor (EGFR) activity and junctional tension to inhibit premature TJ complex formation in lower layers, while promoting TJ stability and cortical stiffness in apical layers. In E-cadherin knockout models, occludin—a transmembrane protein essential for TJs—and its cytosolic connector ZO-1 exhibit a more punctate or discontinuous pattern at cellular interfaces, explaining why TJ barrier function is compromised in the absence of E-cadherin (Rübsam et al., 2017).
Moreover, TJ proteins can influence E-cadherin regulation. For instance, introducing mutated ZO-1 into a ZO-null cell line inhibits the maturation of AJs during epithelial polarization (Ikenouchi et al., 2007). Additionally, overexpression of claudin-1 has been shown to drive the transcriptional downregulation of E-cadherin through the transcriptional repressor ZEB-1 (Singh et al., 2011). In contrast, overexpression of claudin-7 upregulates E-cadherin expression and enhances cell-cell adhesion, whereas E-cadherin expression does not appear to induce an increase in claudin-7 (Lioni et al., 2007).
2.5 E-cadherin and Wnt pathwaysThe Wnt signaling pathways are evolutionarily conserved cellular communication networks that play a key role in both normal physiological and disease states. Several studies have reported that Wnt signaling governs processes such as cell fate determination, differentiation, proliferation, migration, and polarity. The pathway is divided into two main branches: the canonical Wnt/β-catenin pathway, which involves the stabilization and nuclear translocation of β-catenin, and the non-canonical Wnt pathways, such as the planar cell polarity (PCP) pathway, which operate independently of β-catenin (Komiya and Habas, 2008; Katoh, 2017; Flores-Hernández et al., 2020). Of note, E-cadherin/β-catenin membranous interaction and colocalization sequesters β-catenin to the membrane, inhibiting Wnt activation and epithelial-to-mesenchymal transition (EMT) by averting nuclear translocation of β-catenin. The Wnt/β-catenin signaling culminates in the nucleus with the formation of the TCF/LEF complex, initiating the transcription of Wnt target genes. Loss of E-cadherin results in downregulation of membrane β-catenin binding, whereas nuclear mutant β-catenin induces EMT, dysregulating the assembly of TJs and AJs (Kim et al., 2019). Also, E-cadherin/β-catenin interaction maintains low levels of cytoplasmic β-catenin fraction by inhibiting Wnt signaling (Stockinger et al., 2001). In reverse, the absence of Wnt stimulus empowers β-catenin phosphorylation by a destruction complex consisting of APC, Axin, GSK3β, and CK1, which marks β-catenin for degradation by the proteasome (Stamos and Weis, 2013). β-catenin´s growth-inducing transcriptional activity can thus be counteracted by E-cadherin, which in turn induces cell cycle arrest or, more pronouncedly, apoptosis (Stockinger et al., 2001).
2.6 E-cadherin and Hippo pathwayThe Hippo pathway is another evolutionarily conserved signaling network that regulates cell-cell communication and tissue homeostasis across species. It integrates environmental signals, including cellular polarity, contact inhibition, soluble factors, and mechanical stimuli, to regulate key biological processes such as cell proliferation, organ/tissue size, development, and regeneration (Cheng et al., 2020; Ahmad et al., 2022; Fu et al., 2022; Nita and Moroishi, 2024; Zhong et al., 2024). It primarily regulates the phosphorylation of Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) by LATS1/2 kinases at multiple serine residues. This phosphorylation facilitates the binding of 14-3-3 proteins, resulting in the retention of YAP/TAZ in the cytoplasm, preventing their nuclear translocation and transcriptional activity, and potentially leading to their proteolytic degradation in the cytosol (Cheng et al., 2020; Zhong et al., 2024). Upon LATS1/2 inactivation, unphosphorylated YAP/TAZ translocate to the nucleus, where it functions as a transcriptional co-activator by associating with the transcriptional enhanced associate domain (TEAD) transcription factor family (Kaan et al., 2017; He et al., 2021). The resulting YAP/TAZ-TEAD complex facilitates the transcriptional activation of numerous target genes, including those encoding critical junctional proteins such as desmogleins and E-cadherin. Inhibition of YAP–TEAD interactions lead to a substantial decrease in both YAP and phospho-YAP levels, significantly impairing cell–cell junction integrity and resulting in the disassembly of AJs and desmosomes (Ahmad et al., 2022). Kim et al. demonstrated that cell-cell adhesion, mediated by homophilic binding of E-cadherin, contributes to YAP inactivation (Kim et al., 2011). Perturbing the E-cadherin/α-catenin complex reduces YAP phosphorylation and increases YAP nuclear accumulation and activity (Kim et al., 2011; Lamar et al., 2012). Studies have shown that the regulation of Hippo pathway kinases and the sequestration of YAP occur at AJs, where several Hippo pathway components are localized (Pan et al., 2018; Ma et al., 2020; Ahmad et al., 2022).
Several studies have also established a connection between Hippo signaling and cell-cell contact through the regulation of TJs, including ZO proteins (Kaan et al., 2017; Ahmad et al., 2022; Guo et al., 2022). Specifically, AMOTL2, a member of the Angiomotin (AMOT) family of proteins, binds directly to the WW domains of YAP via its PPxY motifs, sequestering YAP at TJs and preventing its nuclear activity. In addition, it has been shown that AMOTL2 interacts with LATS2, permitting the recruitment of upstream Hippo components, such as SAV1, to the junctional complex. The interaction between AMOTL2 and LATS2 also facilitates LATS2-mediated YAP phosphorylation, cytoplasmic retention, and inactivation (Paramasivam et al., 2011; Zhao et al., 2011). Intriguingly, the scaffolding functions of AMOTL2 have been described beyond YAP and LATS2, including multiple other junctional proteins like ZO-1 and β-catenin, thus contributing to maintenance of TJ integrity and epithelial polarity (Zhao et al., 2011; Kim et al., 2021). Hippo and canonical Wnt have been reported to engage in crosstalk, particularly through the YAP effector; YAP/TAZ has been described as part of the β-catenin destruction complex and can modulate the Wnt/β-catenin response and β-catenin degradation; in Wnt-OFF cells, YAP/TAZ cytoplasmic sequestration as part of the destruction complex, inhibits Wnt/β-catenin signaling in the cytoplasm. Conversely, in nucleus, YAP/TAZ can contribute to β-catenin-mediated transactivation of genes, with the two co-activators complexing and β-catenin/YAP/TAZ/TEAD co-regulating target genes. Finally, YAP can be a Wnt/β-catenin target gene, with its expression being a driver of proliferation in cancer cells (Konsavage and Yochum, 2013; Sileo et al., 2022).
3 E-cadherin regulation in bacterial infectionsE-cadherin is considered the gatekeeper of the epithelial barrier, which stands at the frontline of mechanical and immune defense against pathogens. Given the biological complexity of inflammation in epithelial tissues and the range of its clinical manifestations, mucosae and other membranes play a crucial role as the first line of defense against bacterial invasion (Haq et al., 2019; Yang and Yan, 2021; Chegini et al., 2023). Specifically, E-cadherin has been implicated in microbial invasion and dissemination during infectious diseases which breach the epithelial barrier. Herein, we report the direct E-cadherin-driven interactions with infectious agents (Tables 1, 2) as well as pathogen-induced signaling and expression dysregulation, which are involved in the etiopathogenesis of bacterial infections (Figure 3).
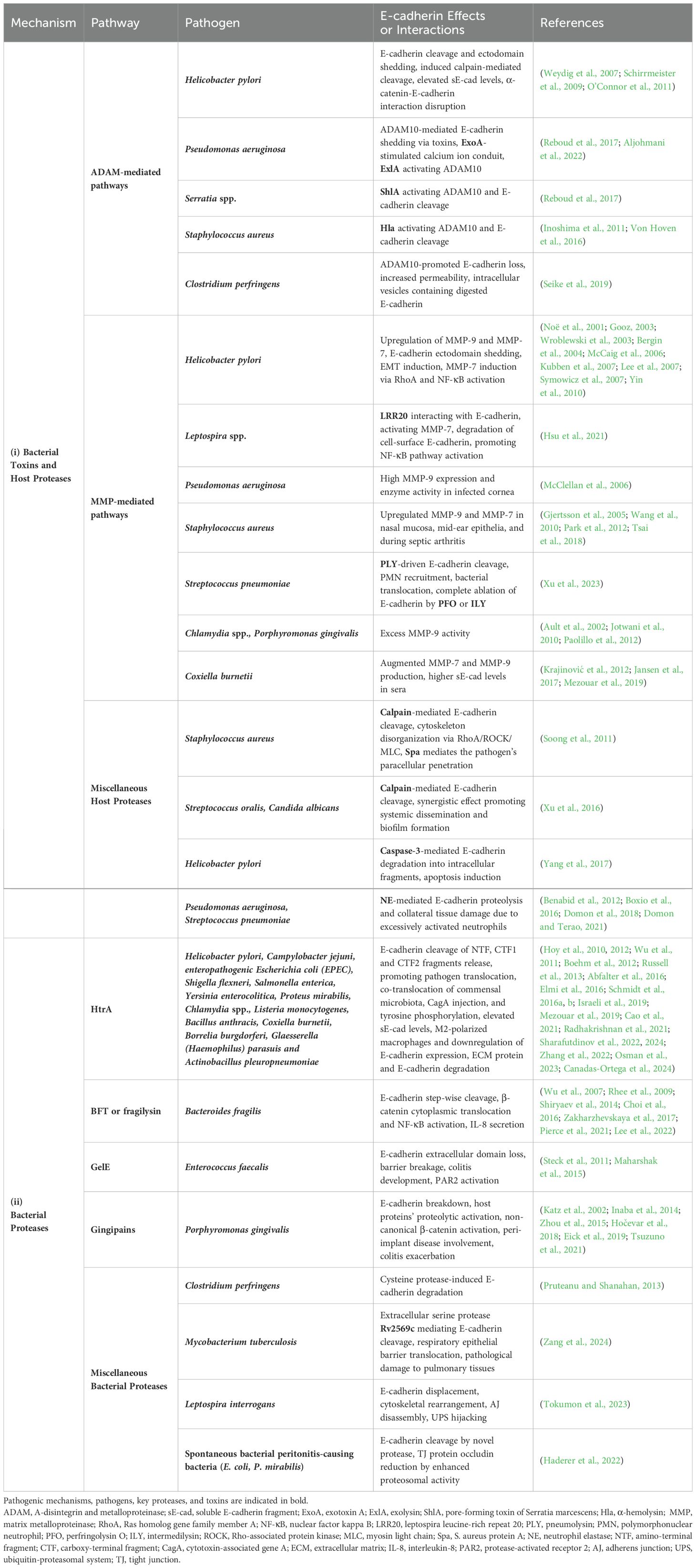
Table 1. Major pathogens, secreted proteases and host sheddases induced by bacterial infection, allow proteolytic degradation of E-cadherin, disruption of the epithelial barrier, and ultimately bacterial invasion and dissemination.
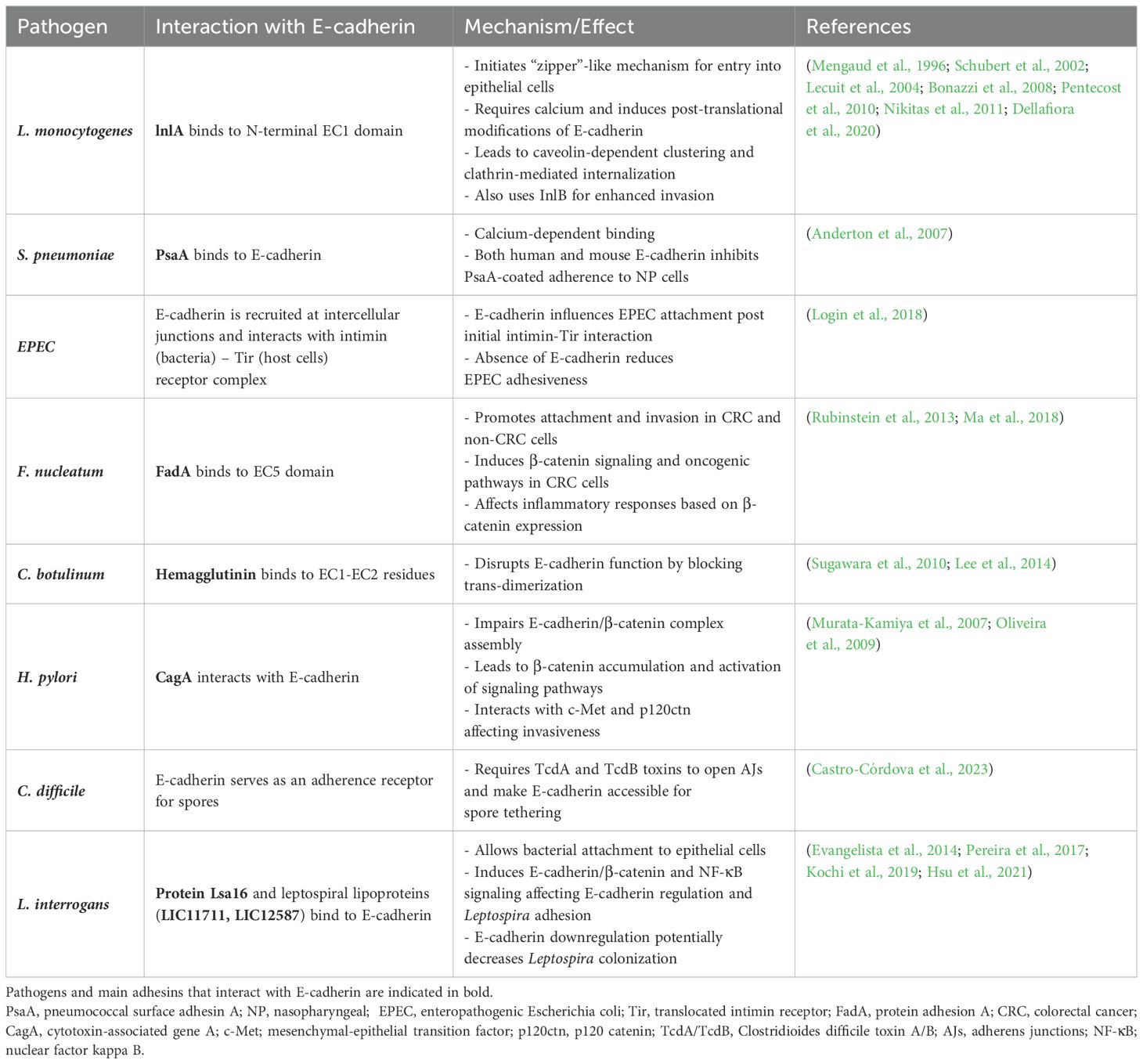
Table 2. Bacterial mechanisms employing E-cadherin as a target receptor for bacterial attachment and entry.
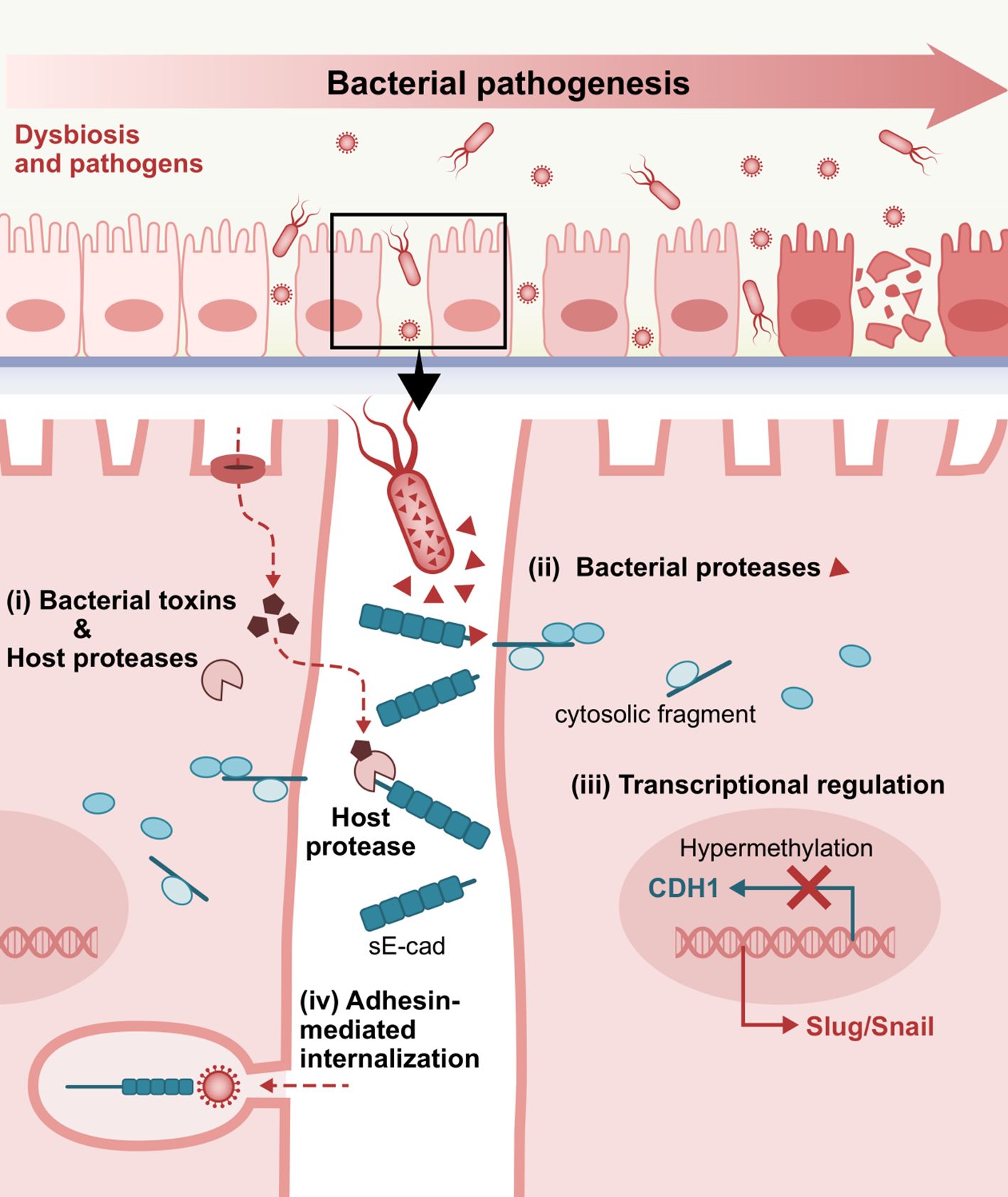
Figure 3. Representative E-cadherin-mediated intercellular interactions that are involved in bacterial pathogenesis. i) Bacterial toxins inducing host proteases, ii) Bacteria-secreted proteases, iii) Dysregulated E-cadherin expression and signaling, and iv) Adhesin-mediated internalization via interactions with the extracellular domain of E-cadherin as a target receptor, play a crucial role in bacterial attachment, invasion into the underlying tissues and consequent establishment and dissemination of infection. sE-cad, soluble E-cadherin fragment.
3.1 Bacterial toxins and pathogen-induced host proteasesE-cadherin cleavage to an 80 kDa soluble fragment is one of the primary mechanisms known to provoke functional loss of E-cadherin. The cleavage of E-cadherin is more commonly attributed to matrix metalloproteinases, including MMP-3 (stromelysin-1), MMP-7 (matrilysin), MMP-9 (gelatinase B or gelatinase type IV), as well as certain ADAMs such as ADAM10 (adamalysin) (Noë et al., 2001; David and Rajasekaran, 2012; Boukhedouni et al., 2020; Tao et al., 2021; Im et al., 2022).
3.1.1 ADAM-mediated pathwaysElevated sE-cad levels have been reported in sera of Helicobacter pylori (H. pylori)-positive patients (O’Connor et al., 2011). H. pylori infection, the causative agent of peptic ulcers and one of the leading risk factors of gastric cancer, was found to trigger significant E-cadherin ectodomain shedding, potentially employing host’s native sheddases, such as ADAM10 or less pronouncedly, ADAM19, as executors (Schirrmeister et al., 2009). Loss of full-length E-cadherin can occur irrespective of H.pylori virulence factor CagA and without transactivating β-catenin transcriptional signaling, while disassembly of AJ complexes rapidly follows disruption of α-catenin-E-cadherin interaction and subsequent disassembly of the E-cadherin/β-catenin/p120ctn complex from the actin cytoskeleton (Weydig et al., 2007). H. pylori can also induce calpain-mediated cleavage, resulting in the production of a 100 kDa truncated E-cadherin form, independent of CagA and VacA, but rather via activation of TLR2 by a putative proteinaceous H. pylori surface component. Cytoplasmic translocation of β-catenin and internalization of E-cadherin ensues, with intracellular redistribution of E-cadherin away from cell-contact sites (O’Connor et al., 2011).
Pseudomonas aeruginosa (P. aeruginosa) infection was recently shown to modulate epithelial permeability by triggering exosomal ADAM10-mediated E-cadherin shedding activity via its secreted toxin repertoire and an Exotoxin A (ExoA)-stimulated calcium ion conduit intracellularly (Aljohmani et al., 2022). Likewise, other pore-forming toxins, such as P. aeruginosa-derived exolysin (ExlA), Serratia marcescens-derived ShlA, and Staphylococcus aureus α-toxin or α-hemolysin (Hla) were also found to drive ADAM10 activation and subsequent cadherin cleavage, through potentiating calcium influx and cell death (Inoshima et al., 2011; Von Hoven et al., 2016; Reboud et al., 2017). In the case of Serratia infection, it has been reported that S. proteamaculans invasion requires full-length E-cadherin, while S. grimesii invasiveness can be promoted by both full-length and truncated E-cadherin. Interestingly, E-cadherin expression was shown to increase and redistribute in cell compartments in response to Serratia infection (Tsaplina et al., 2023).
Clostridium perfringens, which is known to cause food poisoning and gas gangrene, encodes a pore-forming toxin named delta-toxin, which can similarly trigger ADAM10-promoted E-cadherin loss in Caco-2 cells, resulting in increased permeability and fluid accumulation in the ileal loop. With respect to E-cadherin degradation, investigators observed the distribution of digested E-cadherin in intracellular vesicles of shedding cells derived from the damaged intestinal villi as soon as 1h after toxin administration (Seike et al., 2019).
3.1.2 MMP-mediated pathwaysA plethora of proteases extending to members of the MMP family, whose substrates include E-cadherin, such as MMP-9 and MMP-7 (matrilysin) (Noë et al., 2001; Lee et al., 2007; Symowicz et al., 2007), are upregulated in H.pylori-infected gastric epithelial tissues (Gooz, 2003; Wroblewski et al., 2003; Bergin et al., 2004; McCaig et al., 2006); MMP-9 exhibits 19-fold higher activity in infected gastric mucosae compared to uninfected ones and is secreted by gastric macrophages in response to bacteria, while it decreases significantly upon H. pylori eradication (Bergin et al., 2004; Kubben et al., 2007). Adherence of the pathogen induced MMP-7 in AGS cells via RhoA and nuclear factor kappa B (NF-κB) activation (Wroblewski et al., 2003). H. pylori-directed EMT through upregulation of E-cadherin-repressive transcription factors Snail and Slug and gastric microenvironment remodeling contribute to its pathogenicity (McCaig et al., 2006; Yin et al., 2010).
A key example of host-pathogen interactions inducing MMP-mediated degradation of E-cadherin is during leptospirosis. An outer membrane virulence factor, leptospira leucine-rich repeat 20 (LRR20), was shown to interact with E-cadherin and mediate its degradation by activating downstream E-cadherin signaling; LRR20 can promote the nuclear translocation of activated β-catenin, significantly increasing MMP-7 expression in a dose and time-dependent manner. LRR20-induced MMP-7 consequently degrades cell-surface E-cadherin, which in turn promotes NF-κB pathway activation (Hsu et al., 2021).
In P. aeruginosa keratitis, MMP-9 was reported to show high expression and greater enzyme zymography activity in the infected cornea of susceptible B6 mice versus normal cornea of resistant BALB/c mice (McClellan et al., 2006).
MMP-9 was significantly upregulated by S. aureus in infected nasal mucosa and mid-ear epithelia, namely chronic rhinosinusitis and lipoteichoic acid-induced otitis media, respectively (Wang et al., 2010; Park et al., 2012), while S. aureus-induced expression depends on PGE2/IL-6 during infection-associated aortic inflammation (Tsai et al., 2018). Elevated MMP-7 contributes to S. aureus septic arthritis pathogenesis, but interestingly, it also eliminates the increased bacterial burden by enhancing bacterial clearance (Gjertsson et al., 2005).
Pneumolysin’s (PLY) pore-forming activity was shown to be essential for Streptococcus pneumoniae to elicit cleavage and subvert organization of E-cadherin at a MOI of 2, though a putatively induced proteolytic executor that remains to be identified (Xu et al., 2023). This low-dose infection drives the recruitment of polymorphonuclear neutrophils (PMNs) and bacterial translocation in a PLY-dependent manner, even in absence of epithelial detachment, while other pore-forming virulence factors of the cholesterol-dependent cytolysins family, such as perfringolysin O (PFO) or intermedilysin (ILY), resulted in almost complete ablation of E-cadherin, indicating a likely pathogenetic mechanism (Xu et al., 2023). Excess MMP-9 activity has been indicated to participate in the pathogenesis of Chlamydia spp. and P. gingivalis infections (Ault et al., 2002; Jotwani et al., 2010; Paolillo et al., 2012). Coxiella burnetii, the etiologic agent of Q fever, can also manifest with augmented MMP(-7,9) production in the acute and persistent form of infection, along with higher sE-cad serum concentrations (Krajinović et al., 2012; Jansen et al., 2017; Mezouar et al., 2019).
3.1.3 Miscellaneous host proteasesCalcium-dependent, non-lysosomal cysteine proteases named calpains, are also known to mediate occludin and E-cadherin cleavage and can be induced by wild-type S. aureus in an EGFR-dependent manner. S. aureus protein A (Spa) mediates the pathogen’s paracellular penetration into polarized airway epithelial monolayers via tumor necrosis factor (TNF) receptor 1 and EGFR stimulation and consequent RhoA/ROCK/MLC activation that disorganizes cytoskeleton distribution, while calpain activity also facilitates staphylococcal transmigration through the ruptured paracellular junctions (Soong et al., 2011). Augmented calpain-mediated E-cadherin reduction has also been observed as a synergistic effect of Streptococcus oralis and Candida albicans coinfection, promoting their systemic dissemination and pathogenic potential of their biofilms (Xu et al., 2016).
Caspase-3, a protease “executioner” involved in apoptosis, has also been associated with E-cadherin dismantling. Degradation of full-length E-cadherin into 3 intracellular/carboxy-terminal fragments (CTF1, CTF2, CTF3) by H. pylori is reportedly coupled with cleaved-caspase-3 upregulation and induction of gastric epithelial cells’ apoptosis (Yang et al., 2017).
Inflammatory responses triggered during bacterial infections are primarily driven by neutrophils. Neutrophil elastase (NE), a serine protease released by neutrophils at the site of acute lung injury, plays a key role in shaping the proteolytic environment during infections, particularly in PMN-rich pathologies. While NE serves a protective function against pathogens, excessive neutrophil activation and dysregulated NE secretion during bacterial infections can lead to tissue damage. Elevated NE levels have been observed in conditions such as pneumonia caused by Pseudomonas aeruginosa, pneumococcal pneumonia, and bacterial exacerbations of chronic obstructive pulmonary disease (COPD) (Benabid et al., 2012; Domon et al., 2018; Thulborn et al., 2019; Domon and Terao, 2021). In a mouse model of P. aeruginosa H103 pneumonia, significant amounts of active NE were detected in bronchoalveolar lavage (BAL) fluids, alongside an approximately 80 kDa fragment of E-cadherin, indicative of its degradation in the alveolar space. This effect was observed after eliminating the confounding influence of bacterial metalloelastases, suggesting that NE itself contributes to E-cadherin breakdown (Boxio et al., 2016).
3.2 Bacterial proteasesIn addition to bacterial stimulation of the host’s native sheddases, proteases encoded and secreted by pathogens have also been described to catalyze E-cadherin fragmentation, independent of endogenous enzymes.
3.2.1 High-temperature requirement A (HtrA)Full-length 125 kDa E-cadherin was identified as a substrate to the serine protease and periplasmic chaperone HtrA, a caseinolytic active enzyme secreted by H. pylori. The HtrA-mediated cleavage of the extracellular 90 kDa amino-terminal domain (NTF) of E-cadherin results in the release of CTF1 that, upon further processing, yields a soluble 33 kDa CTF2 fragment (Hoy et al., 2010). A 29 kDa E-cad/CTF3 fragment can be produced by caspase-3 cleavage in H. pylori-induced apoptosis of gastric epithelial cells (Yang et al., 2017). HtrA was reported to cleave at the linker regions between the EC domains, with the signature cleavage sites potentially being masked under calcium-binding homophilic homotypic interactions (cis and trans) (Schmidt et al., 2016b, a). HtrA was further characterized as a highly conserved virulence factor among bacterial species, with HtrA-mediated E-cadherin truncation potentially comprising a prominent pathogenic mechanism for Gram-negative gastrointestinal pathogens, including H. pylori, Campylobacter jejuni, enteropathogenic Escherichia coli (EPEC), Shigella flexneri, Salmonella enterica subsp. Enterica (S. Typhimurium), Yersinia enterocolitica, and Proteus mirabilis (Hoy et al., 2012; Abfalter et al., 2016). Of note, HtrA-mediated E-cadherin cleavage properties are limited to DegP and DegQ homologs expressed by Gram-negative pathogens, which harbor different HtrAs combinations (Abfalter et al., 2016). The Hoy group showed that HtrA is expressed mainly as active multimers in H. pylori and C. jejuni -as opposed to monomers in EPEC and S. flexneri- allowing the pathogens to efficiently and rapidly transverse polarized MKN-28 monolayers via the paracellular route (Hoy et al., 2012). In H. pylori infection, HtrA-mediated E-cadherin shedding on the surface of highly polarized gastric epithelial cells, permits CagA injection and tyrosine phosphorylation in the cytosol of non-transformed healthy cells (Canadas-Ortega et al., 2024). In the case of C. jejuni, the transmigration does not confer any drastic reduction in transepithelial electrical resistance (TEER), suggesting that HtrA-directed cell-cell junction opening is executed in a strictly controlled, spatiotemporally restricted manner that enables pathogens to seamlessly cross the intercellular space, whereas this translocation capacity is severely defected in ΔHtrA mutants compared to wild-type bacteria (Boehm et al., 2012). C. jejuni outer membrane vesicles (OMVs) with serine protease activity targeting intestinal epithelial E-cadherin and occludin are thought to deploy HtrA to exercise their cleaving effects (Elmi et al., 2016). Yet, the group of Sharafutdinov and colleagues showed by electron and confocal immunofluorescence microscopy that it is not the soluble purified protease nor the protease in HtrA-containing OMVs, but the C. jejuni surface-bound HtrA that disrupts epithelial cell-cell junctions (Sharafutdinov et al., 2024). Moreover, HtrA-expressing C. jejuni was shown to facilitate co-translocation of commensal microbiota with otherwise weak transmigratory capabilities, such as non-pathogenic E. coli and Lactococcus lactis, which may represent a central mechanism that underpins the pathogenesis of inflammatory bowel disease (IBD) (Sharafutdinov et al., 2022). Additionally, HtrA induction as a proteolytic tool that manipulates host cell machinery has been reported in chlamydial infection (Wu et al., 2011) and in Listeria monocytogenes (Radhakrishnan et al., 2021), while it also plays a role in stress resistance and pathogenicity of Bacillus anthracis (Israeli et al., 2019). However, proof of enhanced E-cadherin degradation was not established in these conditions. In Coxiella burnetii infection, secretion of functional cbHtrA was pinpointed as another plausible mechanistic explanation behind the elevated sE-cad levels found in sera of patients with Q fever (Mezouar et al., 2019; Osman et al., 2023). Indeed, recombinant cbHtrA-treated and C. burnetii-infected BeWo cells released markedly higher sE-cad compared to unstimulated cells, while cbHtrA-exposed macrophages skewed toward M2-polarized interleukin signature which additionally downregulated E-cadherin expression (Osman et al., 2023). Borrelia burgdorferi, the causative agent of Lyme disease, is also endowed with HtrA-mediated cleaving capacity in vitro, allowing host extracellular matrix (ECM) protein and E-cadherin degradation, which is consistent with spirochaetal dissemination findings (Russell et al., 2013). Lastly, E-cadherin ectodomain shedding by HtrA/DegQ virulence factor has lately been described in porcine respiratory pathogens such as Glaesserella (Haemophilus) parasuis and Actinobacillus pleuropneumoniae (Cao et al., 2021; Zhang et al., 2022). Studies have shown that bacterial paracellular transmigration was significantly higher in E-cadherin knock-out, as opposed to the effects of HtrA depletion (Cao et al., 2021).
3.2.2 BFT or fragilysin (FRA)The group of Wu et al. proved that enterotoxigenic Bacteroides fragilis leverages a zinc-dependent metalloprotease toxin termed BFT or fragilysin, that shares homology with eukaryotic MMPs, in order to manifest its virulence through BFT-initiated step-wise cleavage of E-cadherin; extracellular ectodomain shedding (80 kDa) and subsequent proteolytic processing with intracellular fragmentation (i.e., 33 kDa, by presenilin-1/γ-secretase) (Wu et al., 2007). Loss of full-length E-cadherin forces dispersion of E-cadherin-bound β-catenin pool and cytoplasmic localization within 1-3 hours. Upon nuclear translocation (3-24 hours), it activates proliferative signaling via TCF pathway activation and c-myc transcription (Wu et al., 2003). Biologically active BFT, capable of E-cadherin degradation, has been found in OMVs as a bacterial secretory delivery system (Zakharzhevskaya et al., 2017). Fragilysin-catalyzed shedding of intestinal epithelial E-cadherin in vivo has been reported to be implicated in murine colitis onset and early IL-8 secretion (Rhee et al., 2009; Lee et al., 2022). Of note, IL-8 induction due to BTF-mediated E-cadherin cleavage is β-catenin-dependent and requires NF-κB signal activation (Lee et al., 2022). MMP-2 was found to be encoded by the same B. fragilis pathogenicity island, but E-cadherin was not recognized as a cleavage substrate (Shiryaev et al., 2014). BFT in anaerobic bacteremia and sepsis has a similar functional role to ADAM10 in S. aureus sepsis. A clostripain-like B. fragilis protease named fragipain is involved in endogenous BTF activation and secretome generation and can directly or indirectly promote E-cadherin-targeted proteolytic activity (Choi et al., 2016; Pierce et al., 2021).
3.2.3 Gelatinase (GelE)Other microbial metalloproteases impairing full-length E-cadherin have been documented, including a GelE produced by commensal Enterococcus faecalis strains; GelE was shown to trigger loss of extracellular E-cadherin and barrier breakage, contributing to the development of experimental colitis in E. faecalis mono-associated IL-10−/− mice, irrespective of antigen-specific activation of colitogenic CD4+ T cells (Steck et al., 2011). Ex vivo epithelial permeability induction by purified GelE appears to require PAR2 activation, while human fecal supernatants from ulcerative colitis (UC) patients can enhance colonic epithelial permeability in wild-type mice, while the effects were lower in PAR2−/− mice (Maharshak et al., 2015).
3.2.4 GingipainsPorphyromonas gingivalis, an established pathogen in adult periodontal disease, is known to secrete three cysteine proteases known as gingipains (HRgpA, RgpB, and Kgp). Gingipains are believed to account for the breakdown of E-cadherin by P. gingivalis, with Kgp being the major degradative effector (Katz et al., 2002). A plethora of other host proteins’ processing has been ascribed to gingipains, including proMMP-9 (Inaba et al., 2014; Hočevar et al., 2018), while β-catenin can also undergo proteolytic activation attributed to gingipains, in noncanonical (Wnt-independent) fashion (Zhou et al., 2015). In peri-implant disease (i.e., peri-implant mucositis and peri-implantitis), gingipains can interfere with sulcular epithelium attachment to titanium–zirconium alloy surfaces through their cleaving ability (Eick et al., 2019). In the intestinal epithelium, gingipains are thought to be employed in murine colitis exacerbated by orally administered P. gingivalis (Tsuzuno et al., 2021).
3.2.5 Miscellaneous bacterial proteasesOther putative microbial cysteine proteases with E-cadherin-cleaving activity have been documented; for instance, Clostridium perfringens culture supernatant induced in vitro degradation of recombinant E-cadherin -albeit no host protease activation-, while cysteine protease inhibitors completely extinguished the proteolytic effects (Pruteanu and Shanahan, 2013).
An extracellular serine protease of Mycobacterium tuberculosis named Rv2569c was recently shown to cleave E-cadherin; M. tuberculosis Rv2569c allowed the bacteria to translocate through the respiratory epithelial barrier in vivo and confer pathological damage to murine pulmonary tissues, promoting colonization and systemic dissemination (Zang et al., 2024).
Leptospira interrogans, etiological agent of leptospirosis, one of the most significant zoonoses globally, is known to displace E-cadherin from the membrane and drive cytoskeletal rearrangement and AJ disassembly by hijacking the host cells’ ubiquitinin-proteasomal system (UPS) and/or lysosomal degradation pathways. Tokumon and co-workers found that L. interrogans specifically triggers E-cadherin endocytosis by mislocalization and degradation of the p120ctn sub-family proteins (p0071 and p120ctn) that interact with the juxtamembrane domain of E-cadherin, through induction of an unidentified protease inhibited by Z-VAD-FMK (Tokumon et al., 2023). The UPS hijacking could also be involved in the degradation of other modulators of cell-cell junctions and cytoskeletal dynamics such as Rho GTPases including Rac1, Cdc42, and RhoA proteins (Tokumon et al., 2023).
Interestingly, a study by Haderer and colleagues investigating the bacterial-to-cell effects in spontaneous bacterial peritonitis (SBP) found that stimulation with E. coli and P. mirabilis led to the cleavage of E-cadherin through a novel bacterial protease activity. In contrast, intestinal bacteria induced the downregulation of the TJ protein occludin via enhancing endogenous proteasomal degradation in colonic epithelial cells (Haderer et al., 2022).
3.3 Transcriptional regulation of E-cadherinBacterial pathogens can seemingly affect E-cadherin expression on a transcriptional level as well as subvert epigenetic alterations that lead to junctional disturbances. P. gingivalis-lipopolysaccharide (LPS) substantially reduced E-cadherin protein expression in epi-4 cells compared to no P. gingivalis-LPS challenge (Abe-Yutori et al., 2017). This expression pattern has been demonstrated in chronic periodontitis subjects, showing a statistically significant decrease in E-cadherin levels compared to healthy individuals, which inversely correlated with K19 increase (Nagarakanti et al., 2007). Semiquantitative immunohistochemical analysis of tissue samples detected a statistically significant reduction in staining intensity from the external oral epithelium, through the gingival sulcus, to the junctional epithelium of clinically healthy gingiva, with the most marked decrease seen in the pathological lining of the pocket epithelium (Ye et al., 2000). In murine gingivitis epithelia, noticeably decreased E-cadherin expression was observed under the inflamed condition on a protein and mRNA level. This was inversely associated with induction of pyroptosis, namely programmed cell death triggered by caspase-1 activation, where caspase-1 and E-cadherin were inversely correlated (Li et al., 2021).
Clostridium perfringens beta2 (CPB2) toxin was shown to confer intestinal epithelial barrier injury in porcine IPEC-J2 cells treated with 20 μg/mL rCPB2 by considerably restricting claudin-1 and E-cadherin mRNA and protein expression levels (Gao et al., 2020). In a transcriptomic analysis of human trophoblast cells (BeWo), many junctional protein genes were recognized as differentially expressed in response to E. faecalis infection, including E-cadherin, which was found significantly downregulated (Tan et al., 2018). E-cadherin transcripts were measured to be progressively inactivated over time in Shigella dysenteriae-infected HT29 cells, with ensuing β-catenin cytoplasmic translocation (Raja et al., 2012).
CDH1 promoter hypermethylation of CpG islands is one of the most common epigenetic patterns that transcriptionally suppress E-cadherin expression. This epigenetic modification is widely considered to have a greater frequency in H. pylori chronic gastritis and constitutes an established early event in gastric carcinogenesis (Chan, 2003; Kang et al., 2003; Liu et al., 2005). In a study, methylation density in gastric body and antral mucosae obtained from H. pylori-positive gastritis patients was approximately 10-fold higher compared to H. pylori-negative patients. The study showed that host inflammatory cytokines and growth factors -including TNF-α, MG132 (ROS), and EGF in response to the infection mediate aberrant E-cadherin methylation and DNA methyltransferase (DNMT) activity in vitro (Miyazaki et al., 2007). IL-1β-stimulated NF-κB cascade activation and DNMT induction via NO production is another compelling transcriptional system engaged in H. pylori-associated hypermethylation status, which conceivably links chronic gastric inflammation and carcinogenesis (Huang et al., 2012). Successful H. pylori eradication therapy notably eliminates methylation effects and results in reversal of prior silencing (Chan, 2006; Leung et al., 2006; Miyazaki et al., 2007), potentially reinstating E-cadherin expression-dependent barrier function. Interestingly, the opportunistic pathogen Acinetobacter baumannii was also found capable of hindering E-cadherin expression through promoter CpG methylation following its nuclear trafficking (Moon et al., 2012). In the pathophysiological course of Chlamydia trachomatis infection, EMT induction also seems to entail methylation increment in the E-cadherin promoter, while upregulation of other mesenchymal markers was not proven to stem from significant epigenetic alterations (Rajić et al., 2017).
3.4 Interactions involving the extracellular domain of E-cadherinGiven that the extracellular part of E-cadherin engages in homotypic and heterotypic interactions to achieve cell aggregation and control cell behavior, bacteria can seize the molecule’s ectodomain as a heterophilic receptor for adherence and uptake by host cells. L. monocytogenes, a food-borne pathogen able of prototypic intracytosolic invasion in non-phagocytic cells, can employ a well-described invasion protein named internalin (lnlA) to interact with the N-terminal EC1 domain via a leucine-rich repeat (LRR) of the bacterial ligand, securing attachment and internalization at the site of the bacterial-epithelial interface (Mengaud et al., 1996; Schubert et al., 2002). Upon specific calcium-requiring anchoring to E-cadherin, L. monocytogenes can initiate lnlA-based and locally constrained entry into the epithelial cells at the sites of bacterial contact without inducing dramatic morphological changes. This type of bacterial ligand-promoted endocytosis more closely resembles the “zipper mechanism” of Yersinia entry but is distinct from Salmonella “trigger” invasion mechanism (Mengaud et al., 1996). Bonazzi et al. showed that lnlA attachment induces sequential E-cadherin post-translational modifications, which are prerequisites for the recruitment of the different components of endocytosis machinery at the bacterial entry site. In this regard, induced Src-mediated phosphorylation and ubiquitination by ubiquitin-ligase Hakai at the juxtamembrane E-cadherin domain were required for caveolin-dependent E-cadherin clustering and clathrin-mediated internalization (Bonazzi et al., 2008). In fetoplacental listeriosis, L. monocytogenes crosses the maternofetal or trophoblastic barrier via heterotypic interaction between accessible syncytiotrophoblast E-cadherin with lnlA, as recapitulated ex vivo in human placental extracts (Lecuit et al., 2004). In the intestinal villi, where E-cadherin is naturally basolateral and secluded from the lumen, L. monocytogenes was shown to exploit transient defects of epithelial polarity and junctional remodeling spots to facilitate penetration. Indeed, multicellular junctions formation in cell extrusion zones of villus tip can function as entry points and enable the pathogen to efficiently reach the apically exposed E-cadherin prior to its dynamin-dependent removal from the cell surface (Pentecost et al., 2010). Apart from extruding apoptotic cells on villi tips or cells located within intestinal epithelial folds, reorganization of apical junctional complexes around goblet cells, which is affected by physical tensions associated with mucus-expelling dynamics, can similarly make E-cadherin luminally accessible. This allows lnlA-initiated rapid transcytosis across intestinal villi vertical axis with ultimate bacterial releas
留言 (0)