
記住我
At the outset of the COVID-19 pandemic, individuals presenting with underlying medical conditions and comorbidities were considered at higher risk for infection with SARS-CoV-2 and the development of complications (Gupta et al., 2023; Russell et al., 2023; Santra et al., 2023; Marušić et al., 2024). The comorbidities reported in the literature that increase risk include obesity, cancer, cardiovascular disease (ischemic heart disease, hypertension, coronary artery disease, congestive cardiac failure, and stroke) diabetes mellitus, obesity, respiratory diseases [chronic obstructive pulmonary disease (COPD), asthma, pulmonary hypertension, and cystic fibrosis], chronic kidney disease, and immunodeficiency (especially people living with HIV [PLWH]) (Gupta et al., 2023; Russell et al., 2023; Santra et al., 2023; Marušić et al., 2024). Interestingly, most investigations on PLWH co-infected with SARS-CoV-2 have reported mild-to-moderate disease with only a few studies finding increased severity and mortality in PLWH (Gatechompol et al., 2021). Risk factors for developing severe SARS-CoV-2 in virally suppressed PLWH are generally similar to HIV-uninfected individuals (Ho et al., 2021).
SARS-CoV-2 is considered a prothrombotic viral infection. Acute coronary syndrome, pulmonary embolism, and deep vein thrombosis are some of the main thromboembolic manifestations observed in patients with COVID-19 (Acherjee et al., 2021). A meta-analysis evaluating the incidence of thrombotic events in patients hospitalized with COVID-19, which included 20,886 patients from 43 studies, reported that venous thromboembolisms occurred in 7.9% (95% CI 5.1–11.2), deep vein thrombosis in 4.1% (95% CI 2.3–6.4%) and pulmonary embolism in 3.5% (95% CI 2.2–5.1%) of patients (Nopp et al., 2020). SARS-CoV-2-induced venous thromboembolism is caused by multiple mechanisms which include i) increased intracellular levels of von Willebrand factor which leads to impairment of endothelial function, ii) exacerbated systemic inflammation initiated by Toll-like receptor activation, and iii) the release of tissue factors by an activated intrinsic coagulation cascade (Acherjee et al., 2021). The inflammatory hyperresponsiveness, defined by increased C-reactive protein (CRP), interleukin (IL)-6, and fibrinogen concentrations, observed during COVID-19 causes an increase in D-dimer levels (Acherjee et al., 2021). Early studies that were done in Wuhan, China, reported a direct correlation between elevated IL-6 and a rise in D-dimer levels, indicating a link between inflammation and the procoagulant state observed during a SARS-CoV-2 infection (Acherjee et al., 2021). SARS-CoV-2 can also infect endothelial cells, resulting in the activation of transcription factors that initiate apoptosis, leading to the release of procoagulant factors (Acherjee et al., 2021). On admission to hospital with COVID-19, patients frequently present not only with coagulopathies but also with hypoxia (Acherjee et al., 2021). Hypoxia responses are orchestrated mainly by hypoxia-inducible factor (HIF).
In normoxic conditions, HIF subunit alpha 1(1α) is recognized by prolyl hydroxylase proteins (PHDs) and is consequently quickly degraded (with a half-life of approximately 5 minutes) by the von Hippel-Lindau tumor suppressor protein (VHL) (Sato and Takeda, 2023; Troise et al., 2023). The VHL protein regulates degradation of HIF-1α via the ubiquitin proteasome pathway by its association with cullin 2, elongins B and C, and ring-box 1 to form an E3 ubiquitin ligase complex that leads to the ubiquitination of HIF-1α (Troise et al., 2023). PHDs act as an oxygen sensitivity system for the HIF pathway before ubiquitination (Troise et al., 2023). During hypoxia, PHDs are inactivated, allowing HIF-1α and HIF-1β to migrate into the nucleus, dimerize, and bind to p300/CREB binding protein (CBP) to form a transcriptional activation complex that binds to the hypoxia response element (HRE) and activates the transcription of target genes (Sato and Takeda, 2023; Troise et al., 2023). Target genes that are upregulated during hypoxic conditions by HIF-1α are involved in multiple biological processes, such as i) angiogenesis [vascular endothelial growth factor A (VEGF-A) and angiopoietin 2 (ANGPT2)] (Marušić et al., 2024), ii) erythropoiesis [Erythropoietin (EPO)], iii) cell metabolism [Glucose Transporter 1 (SLC2A1), Hexokinase 1 (HK1) and Lactate Dehydrogenase A (LDHA)], iv) iron metabolism [Heme Oxygenase 1 (HO1) and transferrin (TF)], and v) apoptosis and cell survival [BCL2Interacting Protein 3 (BNIP3) and insulin-like growth factor-1(IGF1)] (Cheng et al., 2017; Sato and Takeda, 2023; Troise et al., 2023).
VEGF-A is one of the main cytokines associated with angiogenesis (Dabravolski et al., 2022). Notably, increased concentrations of VEGF have been observed in individuals presenting with SARS-CoV-2-associated acute respiratory distress syndrome (ARDS). As such, VEGF-A, together with other endothelial cell adhesion molecules, is considered a biomarker of disease severity, contributing to coagulation dysfunction (Chen L. et al., 2020). Similarly, elevated levels of VEGF-A have been documented in PLWH despite virally suppressive antiretroviral therapy (ART) (Ascherl et al., 1999). Thus, it has been suggested by a few studies that anti-VEGF therapy could be beneficial in treating severe COVID-19 and various clinical trials are still ongoing (Islam et al., 2020; Pang et al., 2021; Patel et al., 2021; Blumberg et al., 2022; Fanning et al., 2022; ClinicalTrials.gov, 2024a; ClinicalTrials.gov, 2024b; ClinicalTrials.gov, 2024c; ClinicalTrials.gov, 2024d; ClinicalTrials.gov, 2024e).
Van der Mescht et al. recently reported significantly elevated levels of VEGF in PLWH co-infected with SARS-CoV-2 compared to those infected with either HIV or SARS-CoV-2 alone (van der Mescht et al., 2023). Interestingly, increased VEGF concentrations were only observed in PLWH with virally suppressed HIV (van der Mescht et al., 2023). The findings of this study suggest that PLWH co-infected with SARS-CoV-2 present with less hypoxia than those without HIV, alluding to an attenuation of the association between hypoxia and elevated levels of VEGF in PLWH (van der Mescht et al., 2023).
In this review article, the role of VEGF-A in the pathogenesis of SARS-CoV-2 and HIV infection is explored, as well as the implications for therapeutic strategies.
2 Vascular homeostasisThe vascular endothelium controls functions such as inflammation, vascular remodeling, cell adhesion, and coagulation (Kamtchum-Tatuene et al., 2019). Endothelial homeostasis is the balance and proper functioning of endothelial cells, which line blood vessels and are crucial for vascular health. This balance is maintained through various mechanisms, including the regulation of vascular tone via nitric oxide (NO) and endothelin, the selective barrier function controlling molecule passage, and the promotion of angiogenesis for new blood vessel formation (Incalza et al., 2018; Peng et al., 2019). Key pathways involved include the NO pathway for vasodilation, the Wnt/β-catenin pathway for cell regulation, the VEGF pathway for angiogenesis, and the TGF-β pathway for vascular integrity (Figure 1) (Walshe et al., 2009; van Meeteren and Ten Dijke, 2012; Tran et al., 2016; Janaszak-Jasiecka et al., 2023).
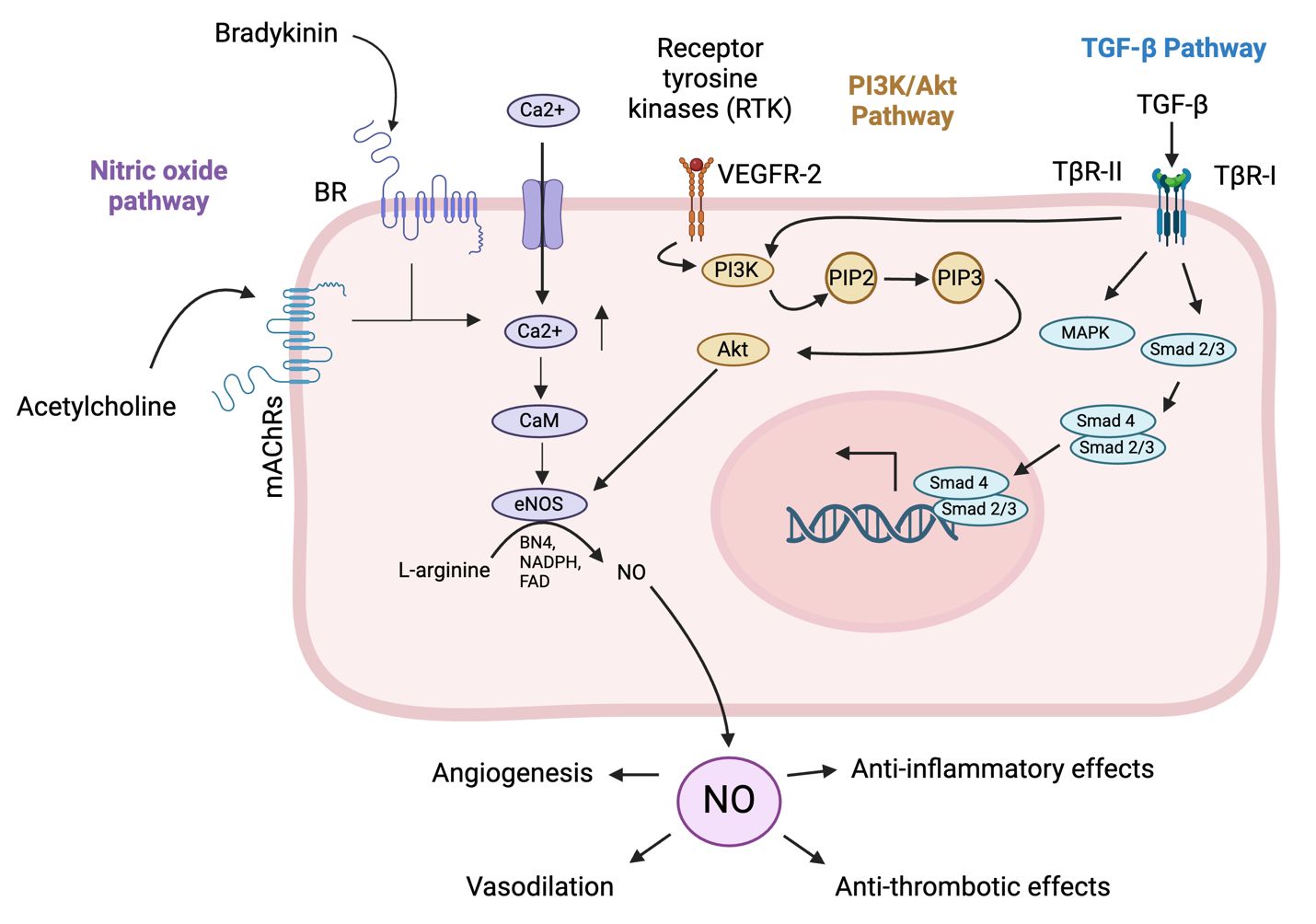
Figure 1. Pathways involved in endothelial cell homeostasis. The NO pathway cascade in endothelial cells begins with the activation of endothelial nitric oxide synthase (eNOS) by stimuli such as shear stress, hormones, and inflammatory cytokines (Durán et al., 2010). Activated eNOS converts L-arginine to nitric oxide (NO) and L-citrulline, requiring co-factors such as tetrahydrobiopterin (BH4) and calcium/calmodulin (Durán et al., 2010). NO then diffuses into adjacent vascular smooth muscle cells (VSMCs) and activates soluble guanylate cyclase (sGC), which converts guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP) (Durán et al., 2010). Increased cGMP levels activate protein kinase G (PKG), leading to the relaxation of VSMCs and vasodilation (Janaszak-Jasiecka et al., 2023). The functional outcomes include vasodilation, anti-thrombotic effects by inhibiting platelet aggregation, and anti-inflammatory effects by modulating adhesion molecule expression on endothelial cells (Durán et al., 2010; Janaszak-Jasiecka et al., 2023). The phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway in endothelial cells begins with the binding of extracellular signals like growth factors such as vascular endothelial growth factor (VEGF) or insulin to receptor tyrosine kinases (RTKs) on the cell surface, leading to RTK autophosphorylation (Zhao et al., 2021). This creates docking sites for PI3K, which then phosphorylates phosphatidylinositol (3,4,5)-trisphosphate (PIP)2 to generate PIP3 (Zhao et al., 2021). PIP3 recruits Akt, with activated Akt in turn phosphorylating downstream targets, promoting cell survival by inactivating pro-apoptotic factors, enhancing protein synthesis via mechanistic Target Of Rapamycin (mTOR) activation, increasing glucose uptake, and boosting NO production for angiogenesis (Zhao et al., 2021). The pathway supports endothelial cell survival, growth, angiogenesis, and metabolic regulation, crucial for vascular integrity and function. The transforming growth factor beta (TGF-β) pathway in endothelial cells begins with the binding of TGF-β ligands to their receptors, transforming growth factor beta receptor (TβR)-II and TβR-I (Huang and Chen, 2012). Upon ligand binding, TβR-II phosphorylates and activates TβR-I, which activates Smad1/5/8, while activin receptor-like kinase 5 (ALK5) activates Smad2/3 (Huang and Chen, 2012). Smad’s form complexes with Smad4 and translocate to the nucleus to regulate gene transcription (Tzavlaki and Moustakas, 2020). Additionally, TGF-β can activate non-Smad pathways such as MAPK, PI3K/Akt, and Rho-like GTPase, influencing cell proliferation, survival, and migration (Huang and Chen, 2012). The functional outcomes include regulating endothelial cell growth and differentiation, maintaining vascular homeostasis, and promoting angiogenesis for wound healing and tissue regeneration. (Created in Biorender).
Different signals can influence the homeostasis of vascular endothelial cells (Chien, 2007). For instance, mechanical stimuli (resulting from circulatory pressure and flow) regulate vascular endothelial function by initiating mechanosensors and signaling pathways, as well as protein and gene expression (Chien, 2007). Mechanical stimuli with an explicit direction lead to transient molecular signaling of proliferative and pro-inflammatory pathways (Chien, 2007). On depletion or removal of these stimuli, the proliferative and pro-inflammatory pathways are downregulated (Chien, 2007). On the other hand, mechanical stimuli without an explicit direction can cause sustained signaling of these pathways, leading to vascular endothelial cell remodeling in order to reduce intracellular stress, as well as changes in vascular endothelial cell signaling to increase tolerance (Chien, 2007). An example of this is the initiation of glycolysis in vascular endothelial cells to promote angiogenesis and vessel sprouting (Lopaschuk et al., 2022). Another example is vascular endothelial cell proliferation secondary to the inhibition of mitochondrial respiration (Lopaschuk et al., 2022). Ultimately, all changes brought about by these mechanisms are balanced by feedback control mechanisms to preserve vascular homeostasis (Chien, 2007).
A healthy endothelium releases NO in response to thrombin secreted by aggregated platelets, which relaxes smooth muscle cells, increasing blood flow, and inhibiting coagulation (Vanhoutte et al., 2017). Shear stress also increases NO release through mechanisms such as bradykinin production in the human coronary circulation (Vanhoutte et al., 2017). The classical definition of endothelial dysfunction is the loss of NO production (Vanhoutte et al., 2017). A reduction of the amount of NO released by endothelial cells when the production of endothelial-derived contracting factor (EDCF) is enhanced is the hallmark of the initiation of endothelial dysfunction (Vanhoutte et al., 2017). The early phase of endothelial dysfunction involves cell activation, higher vascular permeability, and increased expression of adhesion molecules (Gimbrone and García-Cardeña, 2016). NO has a protective effect on the endothelium by preventing abnormal constriction (vasospasm) (Vanhoutte et al., 2017). It also inhibits the aggregation of platelets and the expression of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), which brings about the adhesion and subsequent penetration of immune cells, as well as the release of vasoconstrictor and mitogenic peptide endothelin-1 (ET-1) (Vanhoutte et al., 2017). The abovementioned changes can be triggered by risk factors associated with cardiovascular disease such as hypertension, hyperlipidemia, insulin resistance, and hyperglycemia (Gimbrone and García-Cardeña, 2016). When endothelial dysfunction progresses into the late phase, a prothrombotic state ensues which can cause increased platelet production and subsequent platelet aggregation (Gimbrone and García-Cardeña, 2016). Aggregated platelets release powerful vasoconstrictors such as serotonin and thromboxane A2 (Vanhoutte et al., 2017). Consequent oxidative stress during this phase can also potentiate inflammation in the artery walls (Gimbrone and García-Cardeña, 2016). The aforementioned changes that occur during the late phase of endothelial dysfunction lead to endothelial cell senescence and death (Gimbrone and García-Cardeña, 2016).
The resting endothelium is equipped with cytokine receptors that coordinate particle clearance by resident macrophages (Flaumenhaft et al., 2022). The pulmonary capillaries account for approximately half of the vascular system in the body and are under continuous immune surveillance by neutrophils and macrophages (Flaumenhaft et al., 2022). If no infection is present, the pro-inflammatory response pathways, such as nuclear factor kappa light chain enhancer of activated B cells (NF-κB), myeloid differentiation primary response 88 (MyD88), signal transducer and activator of transcription 3 (STAT3), and mitogen-activated protein kinases (MAPK), are inhibited by cytoprotective receptors (Flaumenhaft et al., 2022).
3 Role of VEGF in angiogenesisSteps involved in angiogenesis include enzymatic degradation of the capillary basement membrane, vascular endothelial cell proliferation, directed migration of vascular endothelial cells, tubulogenesis, vessel fusion, vessel pruning, and pericyte stabilization (Felmeden et al., 2003; Adair and Montani, 2010; Castro et al., 2018). Endothelial tip cells guide the maturing capillaries through the extracellular matrix (ECM) towards an angiogenic stimulus (Adair and Montani, 2010). Angiogenesis is initiated by angiotensin (Ang-2) and VEGF through the activation of the MAPK and protein kinase B (Akt) signaling pathways, which lead to the secretion of proteolytic enzymes (matrix metalloproteinases) by filopodia (Felmeden et al., 2003; De Smet et al., 2009; Adair and Montani, 2010; Barillari, 2020; Shi et al., 2023). Filopodia release substantial amounts of proteolytic enzymes that digest a pathway through the extracellular matrix for the development of the capillary sprout (De Smet et al., 2009; Adair and Montani, 2010; Castro et al., 2018). These enzymes cause the detachment of pericytes from vessels, cleave endothelial cell adhesion molecules, and degrade the basement membrane of the cell, thereby releasing the angiogenic molecules contained within the cells (Castro et al., 2018; Barillari, 2020).
VEGF undergoes alternative exon splicing, resulting in multiple isoforms, including VEGF121 (a highly diffusible isoform), VEGF165, VEGF189 (an ECM-bound isoform), and VEGF206 (Apte et al., 2019). The relative proportions of these VEGF-A isoforms vary across different tissues due to their specific roles in the later stages of vascular development and the affinity of various VEGF-A receptors (Mackenzie and Ruhrberg, 2012).
VEGF165, the most physiologically relevant isoform, is also the most highly expressed in tissues. It exhibits characteristics shared by VEGF121 and VEGF189, being partially matrix-bound and partially diffusible (Mackenzie and Ruhrberg, 2012; Apte et al., 2019). The diffusibility of these isoforms are linked to their varying affinity for heparan sulfate proteoglycans (HSPGs) in the ECM, although there is still a lack of genetic or physiological evidence supporting the hypothesis that HSPGs are essential for VEGF-A isoform-induced signaling events (Mackenzie and Ruhrberg, 2012).
VEGF121 has minimal affinity for heparin, whereas VEGF189 and VEGF206 each possess two heparin-binding domains encoded by exons 6 and 7, which anchor the protein to the cell surface or ECM (Apte et al., 2019). VEGF165 contains a single heparin-binding domain, encoded by exon 7, making it partially diffusible and partially ECM-bound (Apte et al., 2019). Proteolytic processing at the carboxyl terminus by enzymes such as matrix metalloproteinase-3 (MMP3) and plasmin can convert ECM-bound peptides into non-heparin-binding, diffusible molecules (Apte et al., 2019). The structures of two fragments of VEGF165 have been determined: the heparin-binding domain (HBD), which consists of 111–165 residues and is named VEGF55 and the receptor-binding domain (RBD), which has between 1 and 110 residues, and is termed VEGF110 (Ye et al., 2021). The structure of the full-length VEGF165 has yet to be determined (Ye et al., 2021).
Less common isoforms, such as VEGF145 and VEGF183, have been identified (Apte et al., 2019). A distinguishing feature of these isoforms is their varying ability to bind heparin (Apte et al., 2019). Recently, several inhibitory isoforms of VEGF, including VEGF165b and VEGF-Ax, have been described (Apte et al., 2019). However, there is some controversy regarding their mechanisms of inhibition (Apte et al., 2019). Notably, VEGF-Ax has been shown to possess pro-angiogenic and pro-permeability properties (Apte et al., 2019).
VEGF-A plays an important role in angiogenesis and is secreted by many types of immune cells, such as macrophages, dendritic cells, activated T-cells and mast cells, and is also found in the α-granules of platelets (Gavalas et al., 2012; Morrell et al., 2014; Li et al., 2016; McHale et al., 2019; Domokos et al., 2023). VEGF-A has two receptors: VEGF receptor (VEGFR)-1 and VEGFR-2. VEGFR-1 has a higher binding affinity for VEGF-A, but the tyrosine kinase activity of this receptor is 10-fold weaker than that of VEGFR-2, implying that it functions as a control receptor during angiogenesis (Talotta, 2022). Because the tyrosine kinase activity of VEGFR-1 is so much weaker, VEGF-A is unable to stimulate the proliferation of cells overexpressing VEGFR-1 (Shibuya, 2006). Thus, VEGFR-1 can act as a positive regulator of angiogenesis by contributing a mild signal towards migration and proliferation (Shibuya, 2006). Indeed, it has been demonstrated in vivo that angiogenesis activated via VEGFR-1 is insignificant (Shibuya, 2006). On the other hand, VEGFR-1 can act as a negative regulator of angiogenesis by sequestering VEGF-A via its ligand-binding domain (Shibuya, 2006).
With respect to macrophage function, VEGF-A enhances the recruitment of these cells (Schnittman et al., 2023). VEGFR-1 is present on the surface of macrophages and monocytes and can cause an adverse immune response upon stimulation (Shibuya, 2006; Ohkubo et al., 2014; Varricchi et al., 2018; Talotta, 2022). The binding of VEGF-A to VEGFR-1 on the surface of monocytes induces migration and chemotaxis of cluster of differentiation (CD)16+ monocytes (Varricchi et al., 2018). VEGFR-1 is more abundantly expressed on the surface of CD16+ (intermediate and non-classical) monocytes than CD16- (classical) monocytes (Varricchi et al., 2018). In monocytes, signaling through VEGFR-1 activates the p38 kinase, ERK 1/2, MAPK, and PI3K/Akt pathways (Tchaikovski et al., 2008). Thus, activation of VEGFR-1 receptors on monocytes and macrophages can increase the secretion of VEGF-A in VEGF-stimulated monocyte-derived macrophages (Ohkubo et al., 2014; Corliss et al., 2016).
The most important complex in the VEGF signaling pathway is VEGF/VEGFR-2 (Jeong et al., 2021). It induces the activation of multiple intracellular signaling pathways, such as the phospholipase C gamma (PLC-γ), protein kinase C (PKC), MAPK, and Akt pathways by means of dimerization (Figure 2) (Barillari, 2020; Jeong et al., 2021; Talotta, 2022). The positive effects of VEGF-A on angiogenesis and the underlying mechanisms of these effects are summarized in Table 1. The filopodia of tip cells abundantly express VEGFR-2; this characteristic allows the cells to sense slight changes in VEGF-A concentrations and align themselves with the VEGF-A concentration gradient (Adair and Montani, 2010). Once enough filopodia on a tip cell have anchored to the substratum, actin filaments within the filopodia pull the tip cell along in the direction of the VEGF-A stimulus (Adair and Montani, 2010). At the same time, endothelial stalk cells proliferate, trailing behind the tip cell, resulting in elongation of the capillary sprout (Adair and Montani, 2010). Vacuoles arise and combine to form a lumen within the stalk cells to become the trunk of the new capillary (Adair and Montani, 2010). At the source of VEGF-A secretion, the tip cells of two or more capillary sprouts converge and fuse to form a continuous lumen, through which oxygen-rich blood can flow (Adair and Montani, 2010).
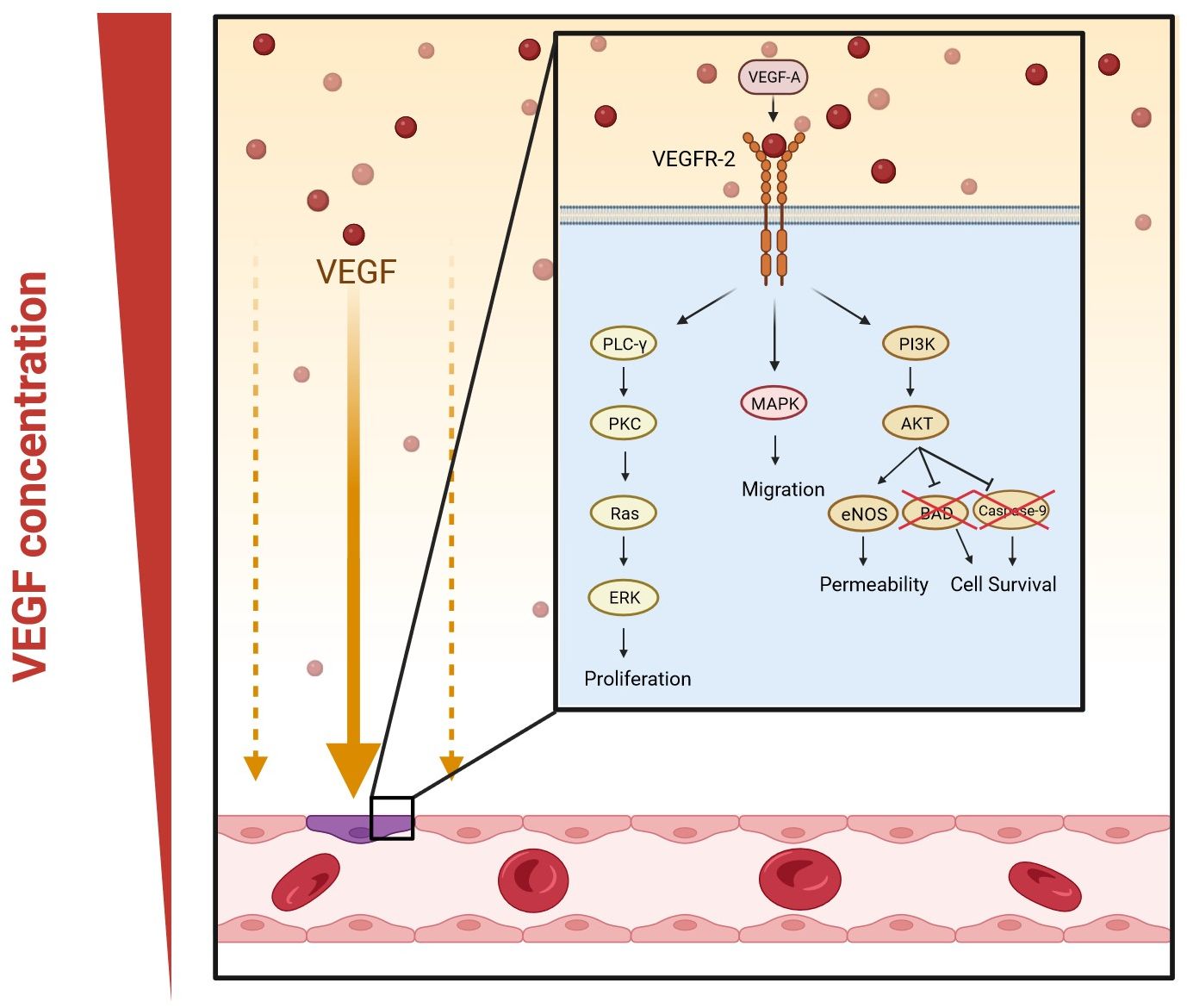
Figure 2. VEGF-A signaling pathways in angiogenesis. Dimerization occurs when VEGF-A binds to VEGFR-2. This activates multiple signaling pathways: phospholipase C gamma (PLC-γ) and protein kinase C (PKC), mitogen-activated protein kinases (MAPK), and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway. The PLC-γ/PKC pathway leads to the activation of extracellular signal-regulated kinase (ERK) and eventually the proliferation of vascular endothelial cells; MAPK increases migration; and the Akt pathway enhances the permeability and survival of the cells by increasing the expression of endothelial nitric oxide synthase (eNOS) and inhibiting Bcl-2-associated death promoter (BAD) and caspase-9. (Created in Biorender).
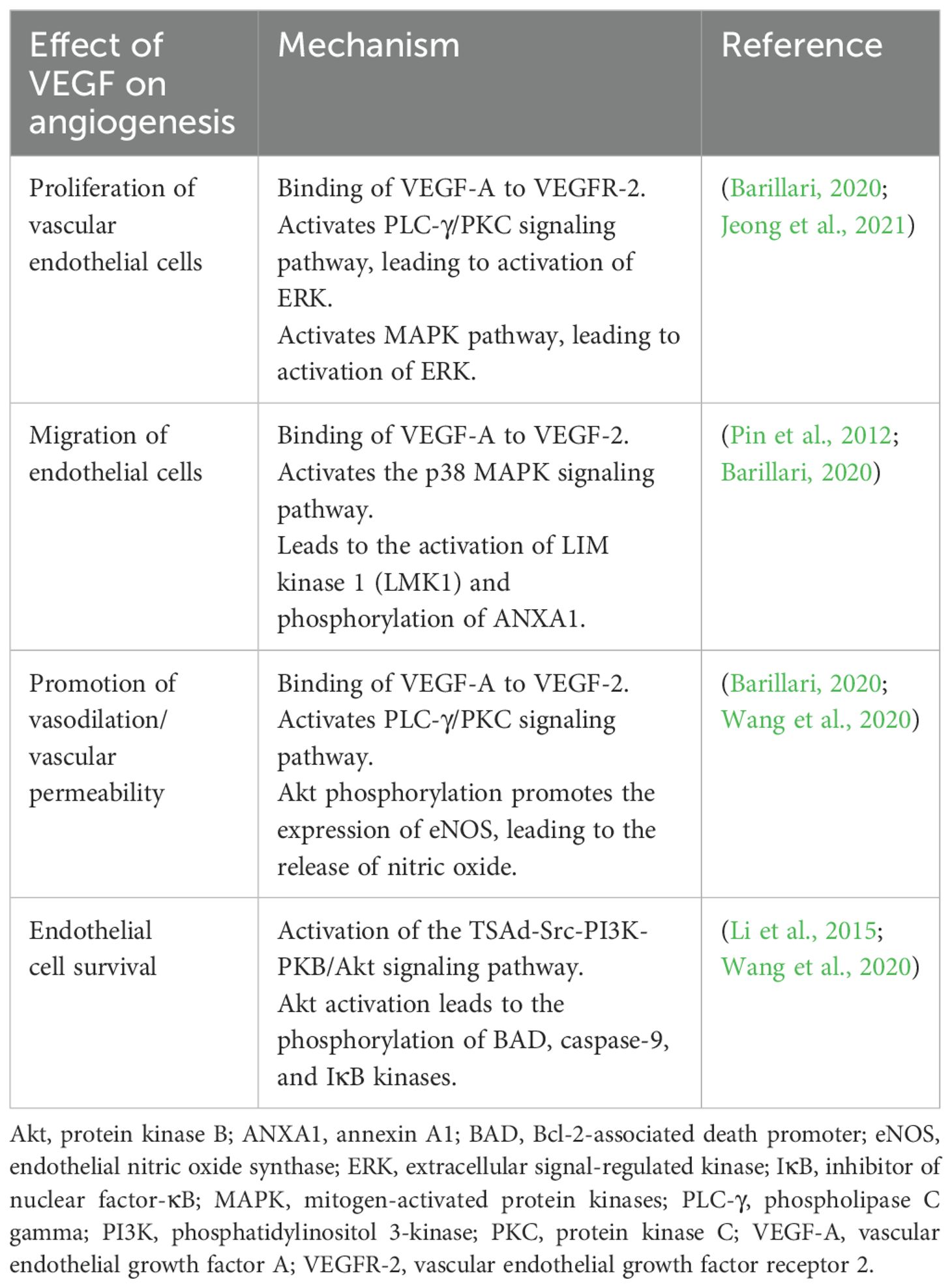
Table 1. Positive effects of VEGF on angiogenesis.
Another key signaling pathway in sprout formation is the Delta-Notch signaling pathway, which is a cell-to-cell signaling system in which cell-bound Delta-like 4 (Dll4) binds to its Notch receptor on the neighboring cell (Adair and Montani, 2010). Once VEGF binds to VEGFR-2, endothelial tip cells secrete Dll4, thus promoting connectivity between proliferating cells by activating the notch receptors on stalk cells (Adair and Montani, 2010; Barillari, 2020). The activation of Notch receptors inhibits the production of VEGFR-2 in stalk cells (Cheng et al., 2017). This is necessary to halt the migration of stalk cells (Adair and Montani, 2010).
Upon completion of angiogenesis, endothelial specialization and rearrangement of vessel connectivity occur (Ouarne et al., 2021). Endothelial cells stabilize the newly formed vessels by recruiting pericytes via the release of platelet-derived growth factor-BB (PDGF-BB) and mechanical signals such as shear stress (Adair and Montani, 2010; Barillari, 2020). These processes are essential for remodeling to occur, such that efficient vascular remodeling leads to the maturation and quiescence of vascular networks (Ouarne et al., 2021). Vascular quiescence occurs when the vascular network is stabilized and stops evolving. This process can be reversed by initiation of angiogenesis through VEGF-A signaling, triggered by different pathological (e.g., viral infections such as SARS-CoV-2) and physiological (e.g., pregnancy) scenarios (Ouarne et al., 2021). Angiopoietin-1 is responsible for the stabilization of endothelial cells and can inhibit vascular leakage (Wada et al., 2013). It has been shown that Ang-1, through the activation of the receptor tyrosine kinase, Tie2, can suppress the expression of genes related to inflammation and coagulation (Wada et al., 2013). Thus, the Ang-Tie2 ligand-receptor system is involved in the regulation and maturation of the endothelium (Wada et al., 2013).
ARDS is defined as a syndrome of acute onset, with bilateral diffuse infiltrates, leading to oxygenation impairment and non-cardiogenic respiratory failure (Vassiliou et al., 2020). The pathophysiology is characterized by alveolar and capillary endothelial damage (Vassiliou et al., 2020). Disruption of the endothelial barrier causes the displacement of macromolecules and fluid into the interstitial and pulmonary air spaces, leading to pulmonary edema (Vassiliou et al., 2020). Hyaline membrane formation in the alveolar walls promotes the release of protein-rich fluid and neutrophils into the alveolar space (Vassiliou et al., 2020). Transport of molecules can either occur transcellularly through endothelial cells or paracellularly via inter-endothelial junctions (Vassiliou et al., 2020). During infection, the endothelium becomes leaky and inflamed (Vassiliou et al., 2020). These changes occur to the microvascular endothelial structure to allow immune cells (innate and adaptive) to cross the barrier and reach the site of infection (Vassiliou et al., 2020). However, under overwhelming pathological conditions, this can lead to the development of ARDS. Critical host responses that occur during ARDS include inflammation (the release of pro- and anti-inflammatory cytokines, which can exacerbate organ damage through endothelial injury), coagulation, and fibrinolysis (Vassiliou et al., 2020). Elevated activation of coagulation in conjunction with dysfunction of the anti-coagulant mechanisms are contributors to, and consequences of, ongoing lung injury (Vassiliou et al., 2020). Once lung injury occurs, endothelial homeostasis is disrupted, initiating a cascade of vascular events, such as the activation of extravascular receptors that seal off the damage by inhibiting fibrinolysis and increasing coagulation (Vassiliou et al., 2020).
VEGF-A can also have a protective role in ARDS (Ricard et al., 2021). VEGF-A is present in the alveolar space in healthy individuals and helps to maintain alveolar function (Ricard et al., 2021). Primary human type 2 alveolar epithelial cells express VEGF-A, which under normal conditions acts as a mitogen and stimulant of the alveolar epithelium (Madureira and Soares, 2021). Polymorphisms associated with lower plasma levels of VEGF have been reported in patients with ARDS (Medford et al., 2005; Medford et al., 2009). In addition, VEGFR-2 blockers can cause alveolar apoptosis and emphysema in adults, suggesting that VEGF-A has a pneumotropic role (Madureira and Soares, 2021).
The development of a healthy vasculature depends on VEGF-A (Adair and Montani, 2010). If VEGF-A levels are reduced by 50% during embryonic development, this proves fatal due to consequent vascular defects (Adair and Montani, 2010). On the other hand, continuously elevated levels of VEGF-A, as seen in tumors due to the overproduction of tip cells, cause the formation of disorganized vasculature (Adair and Montani, 2010). Some of the pathophysiological conditions and the underlying mechanisms associated with VEGF are summarized in Table 2.
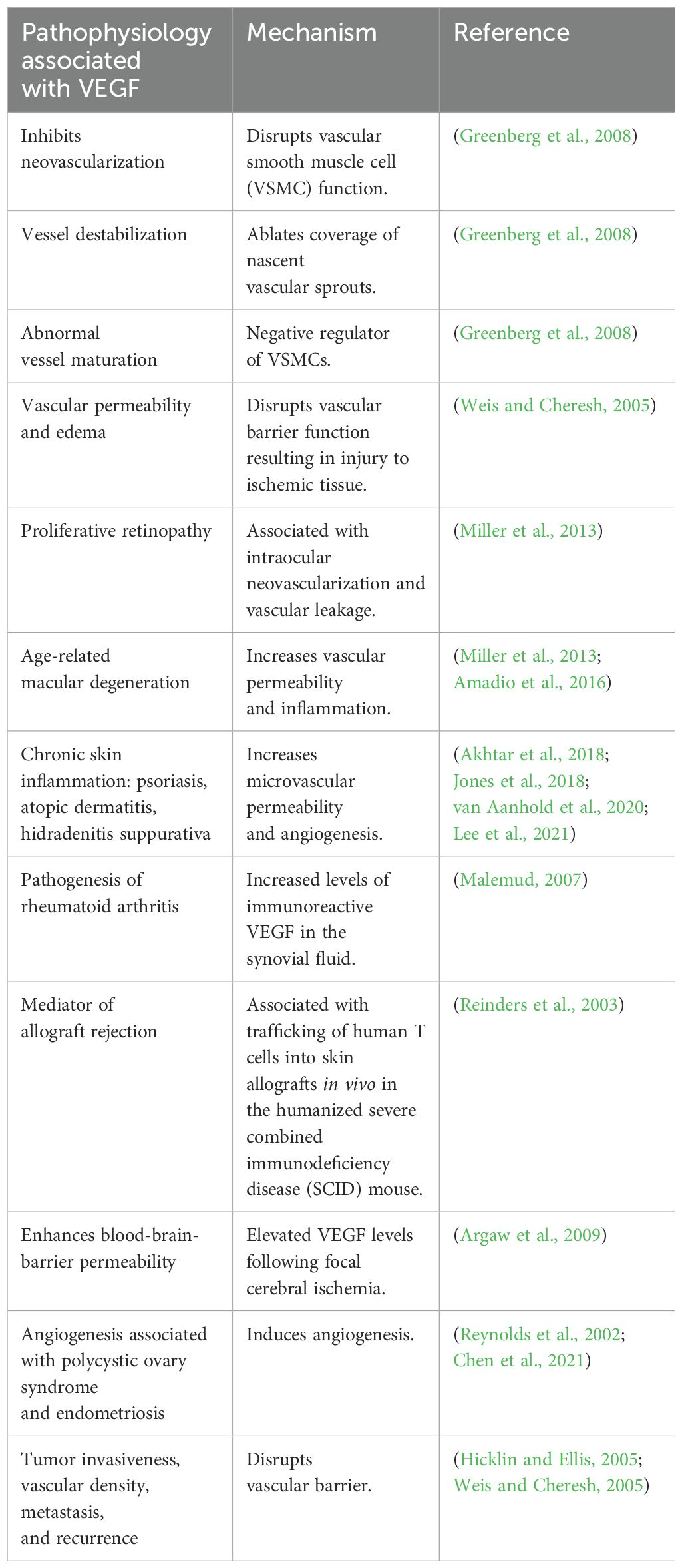
Table 2. Pathophysiological conditions associated with VEGF and the mechanisms underlying these effects.
4 The interplay between VEGF-A and neuropilin-1Neuropilin-1 (NRP-1) is a receptor found on endothelial cells that engages in cell signaling pathways involved in cell migration (Dabravolski et al., 2022). If VEGF-A is secreted close to blood vessels, it upregulates the expression of NRP-1 on the cell surface, creating a positive feedback loop by increasing the endothelial sensitivity to VEGF-A (Talotta, 2022). NRP-1 is most abundantly expressed by mesenchymal stem cells, endothelial cells, and vascular smooth muscle cells (Mayi et al., 2021). Other cells that express NRP-1 are CD8+ T cells, regulatory T cells, macrophages, and neurons (Mayi et al., 2021). PDGF and arterial injury both upregulate the expression of NRP-1 (Mayi et al., 2021). Although NRP-1 induces angiogenesis without the involvement of VEGF-A, the VEGF pathway cannot function properly without NRP-1, underscoring the critical role of NRP-1 in the functioning of the vascular system (Mayi et al., 2021).
Different isoforms of NRP-1 can be produced due to alternative splicing (Talotta, 2022). Some of these isoforms are soluble and can function as decoy receptors (Talotta, 2022). A review by Roy et al. showed that S12NRP-1 acts as a decoy, which inhibits VEGF165 binding to NRP-1 since it contains the a and b domains, but lacks the transmembrane and cytosolic residues (Roy et al., 2017). The b1 coagulation factor domain of NRP-1 and NRP-2 contains a conserved cleft, which is best suited for interacting with a C-terminal arginine necessary for ligand binding (Parker et al., 2012). All VEGF family members contain a C-terminal arginine (Parker et al., 2012). While NRP-1 is the functional receptor for VEGF-A, NRP-2 is the functional receptor for VEGF-C (Parker et al., 2012). The b1 domain, which also interacts with furin-cleaved ligands that share a motif with arginine amino acid residues, is common in VEGF-A (Talotta, 2022).
NRP-1 forms a complex with VEGFR-2 in endothelial cells (Mayi et al., 2021; Talotta, 2022). NRP-1 and NRP-2 both act as co-receptors to amplify the binding of VEGF to VEGFR-2 in blood vessels (Talotta, 2022). NRP-1 has strong pro-inflammatory and pro-atherogenic properties by promoting leukocyte trafficking to sites of inflammation (Schnittman et al., 2023). In a murine model, NRP-1+ CD4+ T cells that are highly activated and secrete interferon (IFN)-γ and tumor necrosis factor (TNF)-α, showed increased migration to the aorta, where they were elevated in atherosclerotic plaques (Schnittman et al., 2023). In this context, it is interesting that NRP-1 expression is upregulated by oxidized low-density lipoprotein (LDL) in the mouse model (Schnittman et al., 2023).
Murine models have also revealed an association between upregulated NRP-1 expression and age (Schnittman et al., 2023). Upregulated NRP-1 expression in these models led to inhibition of anti-thrombotic and anti-inflammatory pathways, ultimately leading to platelet and macrophage activation and fibrosis (Schnittman et al., 2023). In a study done in humans, other clinical factors that were associated with elevated NRP-1 include a lower CD4+ T cell count, male sex, and a history of hepatitis C virus infection (Schnittman et al., 2023).
NRP-1 further acts as a signaling ligand for plexin, semaphorins, integrins, and various growth factors, such as PDGF, hepatocyte growth factor (HGF), and TGF-β1 (Mayi et al., 2021). PDGF mediates the hypoxia-induced promotion of proliferation and survival of endothelial cells by initiating receptor dimerization and phosphorylation, leading to the activation of multiple signaling pathways such as Akt/PKB, MAPK/ERK and JAK/STAT (Li et al., 2015; Zou et al., 2022). PDGF further contributes to cell survival by protecting cells against mitochondria-dependent apoptosis through the activation of the Akt/STAT3 signaling pathway (Li et al., 2015). HGF is a multifunctional growth factor involved in anti-inflammatory responses, angiogenesis, and cell proliferation and migration (Kaga et al., 2012). HGF can induce angiogenesis without inducing vascular permeability and inflammation (Kaga et al., 2012). These interactions underscore the contribution of NRP-1 to cell survival, migration, and proliferation (Talotta, 2022).
5 Role of VEGF-A during hypoxia and inflammationDuring hypoxia and inflammation, endothelial cells are stimulated to release HIF-1α, which, via its role as a transcription factor, upregulates genes that mediate adaptive responses to lower oxygen availability, such as those encoding erythropoietin, VEGF-A, PDGF, and smooth muscle mitogen, to compensate for ischemic and hypoxic damage (Korgaonkar et al., 2008; Barillari, 2020; Dabravolski et al., 2022; Palakeel et al., 2022). VEGF-A is the only growth factor that can cause hypoxia-induced angiogenesis (Adair and Montani, 2010). This process is initiated in tissues during hypoxia in order to meet the metabolic needs of parenchymal cells (Adair and Montani, 2010). Under hypoxic conditions, endothelial cells also produce TNF-α, which activates the NF-κB pathway, thus creating the TNF-α-NF-κB-HIF-VEGF-A signaling cascade (Dabravolski et al., 2022). NF-κB activation is one of the most important events involved in triggering inflammation and proliferation (Dabravolski et al., 2022). In this context, VEGF-A can activate NF-κB through the Akt pathway (Figure 3) (Dabravolski et al., 2022).
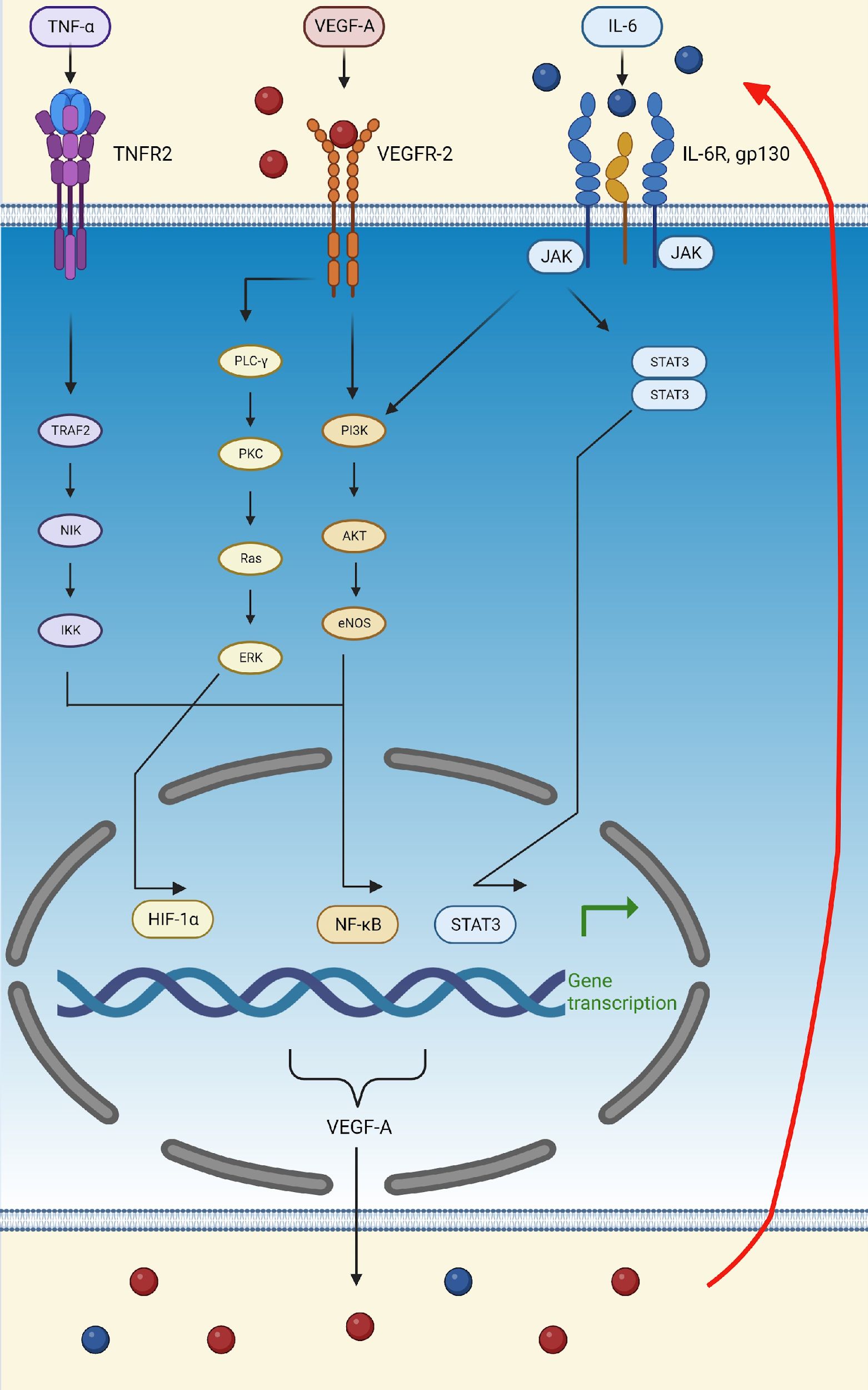
Figure 3. Schematic representation of the TNFα-NF-κB-HIF-VEGF pathway. TNF-α activates the NF-κB transcription factor by binding to TNFR2 and activating TRAF2, leading to the transcription of VEGF-A and other pro-inflammatory molecules, such as IL-6. VEGF-A can then attach to VEGFR-2 and activate the transcription factors, HIF-α and NF-κB, thus further upregulating the production of VEGF-A, IL-6, and TNF-α. Finally, IL-6 can bind to its receptor, IL-6R, activating the PI3K/Akt and STAT3 pathways. The activation of these pathways, initiated by VEGF-A, TNF-α, and IL-6, increases the production of VEGF-A, thus creating a positive feedback loop (indicated by the red arrow). (Created in Biorender). eNOS, endothelial nitric oxide synthase; ERK, extracellular signal-regulated kinase; Gp130, glycoprotein 130; HIF-1α, hypoxia-inducible factor 1 subunit alpha; IκB, inhibitor of nuclear factor-κB; IKK, kinase; IL-6, interleukin; IL-6R, interleukin 6 receptor; JAK, Janus kinase; NF-κB, nuclear factor kappa light chain enhancer of activated B cells; NIK, NF-κB induced kinase; PI3K, phosphatidylinositol 3-kinase; STAT3, signal transducer and activator of transcription 3; TNF-α, tumor necrosis factor-alpha; TNFR2, tumor necrosis factor receptor 2; TRAF2, TNF receptor-associated factor 2; VEGF-A, vascular endothelial growth factor A; VEGFR-2, vascular endothelial growth factor receptor 2.
In addition to TNF-α, lipopolysaccharide (LPS) and reactive oxygen species (ROS) also activate the TNFα-NF-κB-HIF-VEGF-A pathway, thereby upregulating IL-6, activating STAT3, and leading to the secretion of VEGF-A (Dabravolski et al., 2022). In turn, VEGF-A stimulates STAT3 in a positive feedback loop and amplifies its secretion (Dabravolski et al., 2022). Lastly, activation of the Akt pathway supports the feedback loop of the IL-6/STAT3 pathway (Figure 3) (Dabravolski et al., 2022).
Furthermore, VEGF-A can inhibit the inflammatory response, reduce oxidative stress, and promote the dilation and proliferation of lymphatic vessels (Dabravolski et al., 2022). In this context, VEGF-A can induce anti-inflammatory conditions by inhibiting the proliferation and function of cytotoxic T cells and through the recruitment of M2-type macrophages through the expression of C-X-C motif chemokine ligand 12 (CXCL12) (Elias et al., 2013; Li et al., 2015). Activated macrophages can be categorized into two distinct types: M1 macrophages, which play a role in promoting inflammatory responses, and M2 macrophages, which are associated with anti-inflammatory immune reactions (Yunna et al., 2020). High concentrations of CXCL12 are associated with some pathological conditions linked to hypoxia and a pro-angiogenic environment (Elias et al., 2013). VEGF-A alone only recruits macrophages, but cannot facilitate macrophage class switching from the M1 to the M2 phenotype (Elias et al., 2013). A study done by Shou et al. demonstrated in vitro that class switching from M1 to M2 phenotype macrophages is promoted by microRNA (miR)-126 via the downregulation of the VEGF-A/Krüppel-like factor 4 (KLF4) signaling pathway (Shou et al., 2023). KLF4 promotes cell proliferation and differentiation of monocytes, polarizing differentiation towards an M2 phenotype (Shou et al., 2023). In an M2-rich environment, the inflammatory response is downregulated, allowing tissue remodeling to occur (Shou et al., 2023).
Protein homeostasis is regulated by the endoplasmic reticulum (ER), which controls protein processing and folding (Wei et al., 2023). ER stress occurs when there is a disruption in the process of protein folding, which leads to an increase in the amount of misfolded proteins in the ER (Wei et al., 2023). HIF-1α is associated with ER stress and promotes apoptosis of alveolar epithelial cells (Luo et al., 2022; Wei et al., 2023). Endothelial cell survival is regulated by VEGF-A signaling under conditions of ER stress via the activation of unfolded protein response mediators through PLC-γ mediated cross-talk with the mechanistic Target Of Rapamycin (mTOR) kinase complex (Dabravolski et al., 2022). Other activated complexes that contribute to the survival effect of VEGF-A on endothelial cells are activating transcription factor 6 and protein kinase R-like ER kinase (Wei et al., 2023).
VEGF-A concentrations are downregulated, and angiogenesis terminates once the tissues receive adequate amounts of oxygen (Barillari, 2020; Talotta, 2022). This downregulation can occur via multiple mechanisms. The expression of VEGF is correlated with lipid levels, thus the TNFα-NF-κB-HIF-VEGF-A signaling cascade can be inhibited by LDL (Dabravolski et al., 2022). In this context, LDL suppresses TNF Receptor superfamily 1A and inhibits the expression of HIFs in endothelial cells, hence downregulating VEGF-A expression (Dabravolski et al., 2022). VEGF-A can decrease the activity of plasma lipoprotein lipase, which results in the accumulation of triglycerides in large protein granules and LDL (Dabravolski et al., 2022). Additionally, a cell cycle transcription factor, E2F Transcription Factor 1, can inhibit VEGF expression in a p53-dependent manner (Dabravolski et al., 2022), whereas placenta growth factor (PlGF) can inhibit VEGF-A in a p53-independent manner. Both events reduce the neovascularization and cardiac regeneration ability following injury (Dabravolski et al., 2022). During homeostatic angiogenesis, annexin A1 is a phospholipase A2 inhibitor with anti-inflammatory properties (Dabravolski et al., 2022). Thus, in addition to other mechanisms, the downregulation of annexin A1, which inhibits the activation of STAT3, further suppresses VEGF-A expression (Dabravolski et al., 2022).
As shown in Table 2, VEGF-A plays an important role in both physiological and pathological angiogenesis, the latter of which occurs during ischemic disease, microvascular occlusion, and inflammation (Dabravolski et al., 2022). Forms of pathological angiogenesis that are known to occur in the lungs include intussusceptive and sprouting angiogenesis, the latter occurring in asthma and chronic obstructive pulmonary disease, while intussusceptive angiogenesis and injured endothelium have been associated with SARS-CoV-2 infection (Jeong et al., 2021). Sprouting angiogenesis occurs when interconnected capillaries are formed from an existing vascular network (Ouarne et al., 2021). Intussusceptive angiogenesis, on the other hand, occurs when an intussusceptive pillar is formed, splitting and remodeling a blood vessel (Ouarne et al., 2021). VEGF-A signaling differs in sprouting and intussusceptive angiogenesis, depending on the local concentration of VEGF-A, as well as the location and the underlying pathological processes in lung tissue (Madureira and Soares, 2021). Lower concentrations of VEGF-A lead to intussusceptive angiogenesis, while higher concentrations of this growth factor lead to sprouting angiogenesis (Madureira and Soares, 2021). The transition between normal to abnormal angiogenesis depends on the concentrations and activities of PDGF-BB and VEGF-A (Meini et al., 2020). PDGF-BB recruits pericytes and thus acts as a regulator by stabilizing the vasculature, thereby preventing abnormal angiogenesis caused by the overexpression of VEGF-A, while a reduction in VEGF-A expression is associated with vascular tree regression by intussusceptive vascular pruning (Meini et al., 2020). Thus, the inhibition of VEGF-A is suggested to lead to vessel normalization by regulating intussusceptive angiogenesis (Meini et al., 2020).
6 VEGF-A upregulation in viral infections and its possible therapeutic implicationsThe NRP-1/VEGF-A axis has been implicated in the pathogenesis of several viral infections, such as those caused by human T cell lymphotropic virus type 1 (HTLV-1) and Epstein-Barr virus (EBV) (Talotta, 2022). The HTLV-1 envelope protein can mimic VEGF-A, promoting viral entry into cells expressing surface NRP-1 (Talotta, 2022). Knockdown of NRP1 has been shown to suppress EBV infection while increasing NRP1 expression has been shown to enhance EBV infection in human nasopharyngeal epithelial cells (Mayi et al., 2021). The two viral infections in which the NRP-1/VEGF-A axis has been implicated most strongly are, however, SARS-CoV-2 and HIV.
6.1 SARS-CoV-2 infectionSARS-CoV-2 is reported to enter cells in multiple ways, including via the angiotensin-converting enzyme 2 (ACE-2) receptor and NRP-1 (Ascherl et al., 1999; Shibuya, 2006; Cheng et al., 2017; Gatechompol et al., 2021; Talotta, 2022; Troise et al., 2023). SARS-CoV-2 has the highest affinity for entry via the ACE-2 receptor, which is expressed in vascular endothelial and respiratory cells, as well as on the surface of many types of immune cells, such as monocytes (Osman et al., 2021) and macrophages (Knoll et al., 2021), as well as in the heart (Romanowska-Kocejko et al., 2022), ileum (Burgueno et al., 2020), kidneys (Akpoviroro et al., 2024), bladder (Lin et al., 2021) and lungs (Li et al., 2020; Gatechompol et al., 2021; Rohlfing et al., 2021).
SARS-CoV-2 binds to the membrane-bound ACE-2 receptor to enter a cell. The strong binding affinity of SARS-CoV-2 to ACE-2 has made COVID-19 a greater threat than previous SARS viruses (Sodhi et al., 2022). This increased binding affinity is due to five amino acid changes (Asn501, Gln493, Leu455, Phe486, and Ser494), which lead to stronger hydrophobic interactions (Sodhi et al., 2022). Among these, Asn501 and Gln493 are the most crucial for van der Waals interactions and hydrogen bonding (Sodhi et al., 2022). The RBD of the viral spike (S) protein of SARS-CoV-2 interacts with the metallopeptidase domain of ACE-2, causing a structural change in the S protein that reveals cleavage sites at the S1/S2 regions (Sodhi et al., 2022).
Before entry, the S protein undergoes priming by the serine endopeptidase, transmembrane serine protease 2 (TMPRSS2), and the cysteine proteases, cathepsin B and L (CatB/L) (Sodhi et al., 2022). TMPRSS2 cleaves the S protein at subunit 1 and 2 sites and the S2 site, allowing the fusion of cellular and viral membranes (Sodhi et al., 2022). Once the S1 subunit binds to ACE-2, it promotes the cleavage of the ACE-2 ectodomain by the protease, A disintegrin and metalloproteinase 17 (ADAM-17), and the intracellular C-terminal domain by TMPRSS2, facilitating SARS-CoV-2 entry (Sodhi et al., 2022). ACE-2 is internalized along with the viral particles into endosomes (Sodhi et al., 2022).
The S protein not only gains entry through the ACE-2 receptor but also interferes with angiogenesis (Talotta, 2022). The ACE-2 receptor downregulates the expression of VEGF-A by preventing the phosphorylation of ERK-2 (Talotta, 2022). By occupying the binding site for the ACE-2 molecule, the S protein interferes with the control of ACE-2 over VEGF-A synthesis, thus increasing the levels of VEGF-A (Talotta, 2022).
Inflammation seen in COVID-19 starts once the virus invades alveolar lung epithelial cells (Silva et al., 2023). After infection, it leads to excessive activation of the ACE/Ang-2/angiotensin type 1 receptor (AT1R) axis, which, in turn, results in these cells being engulfed by antigen presenting cells, both activating and inducing innate and adaptive immune responses (Silva et al., 2023). The immune cells that are then recruited to the site of infection release large quantities of inflammatory mediators such as chemokine (C-C motif) ligand (CCL)7, CCL8, CCL13, CCL17, CD163, CXCL2, CXCL11, IFN-γ, IL-1, IL-2, IL-3, IL-4, IL-6, IL-7, IL-8, IL-9, IL-10, IL-13, IL-15, IL-17, interferon-inducible protein 10 (IP-10), macrophage inflammatory proteins (MIPs), regulated upon activation normal T-cell expressed and secreted (RANTES), soluble tumor necrosis factor receptor-1 (sTNFR1),TNF-α, TNF-β, and TNF-related apoptosis-inducing ligand (TRAIL) (Silva et al., 2023). The release of these numerous inflammatory mediators results in an explosive and uncontrolled immune response with severe symptoms and oxidative stress that has been coined as a cytokine storm (Silva et al., 2023). SARS-CoV-2 impacts both the pulmonary and systemic circulation through the renin-angiotensin system (RAS) pathway, leading to a prothrombotic state with hypercoagulability (Silva et al., 2023). Various immune and endothelial factors, including granzymes and VEGF, together with viral and host proteins such as caspases 6 and 8, intensify the inflammatory process (Silva et al., 2023). Monocytes and neutrophils contribute to the cytokine storm via inflammasome activation (Silva et al., 2023). An elevated neutrophil-to-lymphocyte ratio, activation of inflammasomes, and high levels of extracellular neutrophil traps (NETs) further exacerbate the inflammation (Silva et al., 2023). This ultimately leads to a hyperinflammatory state, which can be defined as an excessive and prolonged inflammatory response by the immune system. Clinical markers used to identify hyperinflammation include the neutrophil/lymphocyte ratio, serum D-dimer, ferritin, LDH and CRP levels (Silva et al., 2023).
The hyperinflammation caused by SARS-CoV-2 infection can upregulate the activation of the VEGF-A/VEGFR-2 pathway, initiating angiogenesis, NO production, vascular permeability, and disruption of endothelial cell junctions (Talotta, 2022). This, in turn, can contribute to interstitial edema and endothelial hyper-permeability (Ricard et al., 2021; Talotta, 2022). High levels of VEGF-A in SARS-CoV-2 infection result in the reorganization of vascular endothelial (VE)-cadherin away from junctional sites, via phosphorylation, leading to the internalization of the protein, thus causing vascular leakage (Flaumenhaft et al., 2022). VEGF-A also promotes vascular permeability and the production of Ang-2 and VE-tyrosine phosphatase, which synergize to block Tie2 signaling (Flaumenhaft et al., 2022). Once the Ang-2 levels exceed the Ang-1 levels, the endothelial barrier is destabilized by the activation of β1-integrins (Flaumenhaft et al., 2022). This, in turn, ultimately leads to the loss of barrier function in postcapillary venules, resulting in fluid extravasation (Flaumenhaft et al., 2022). SARS-CoV-2 can directly infect endothelial cells, causing cell lysis and death, further damaging the barrier integrity (Lambadiari et al., 2022). It has been proposed that deflated alveoli filled with plasma could be attractive to opportunistic bacteria and fungi (Cao, 2021). The cytokine-rich plasma also results in the recruitment of inflammatory cells (such as macrophages), further increasing lung inflammation (Cao, 2021). An increase in VEGF-A could also be a result of endothelial dysfunction, leading to local tissue hypoxia, which triggers a self-perpetuating mechanism (Talotta, 2022). Hypoxia-induced vascular leakage during SARS-CoV-2 infection plays a significant role in the pathogenesis of COVID-19 (Cao, 2021).
High VEGF-A plasma levels are reported during the early stages of SARS-CoV-2 infection (Madureira and Soares, 2021). A study by Josittus et al. that investigated whether VEGF-A levels varied between SARS-CoV-2-positive patients in terms of disease severity found that high VEGF-A levels measured on admission to hospital correlated with mortality (Josuttis et al., 2023). The study concluded that VEGF-A can be used to determine which patients are at risk of developing ARDS, acute kidney injury, or shock (Josuttis et al., 2023). In another study done by Rovas et al., the MYSTIC trial, which analyzed functional and biomarker data from 23 patients admitted to hospital with COVID-19, determined that circulating VEGF-A and ADAMTS13 levels, also known as von Willebrand factor-cleaving protease, as well as sublingual glycocalyx thickness were the main markers that were associated with disease severity and predictive of whether patients will develop ARDS (Rovas et al., 2021).
The elevated VEGF-A levels reported during SARS-CoV-2 infection are indicative of widespread microvascular injury (Talotta, 2022). It is, therefore, likely that high levels of VEGF-A in COVID-19 further increase alveolar damage (Madureira and Soares, 2021). Increased VEGF-A expression in patients with COVID-19 causes impaired pericyte function, which leads to intussusceptive angiogenesis in the lungs (Talotta, 2022). As VEGF-A levels increase, Ang-2 levels also rise. In combination, these two molecules lead to the formation of small perforations in the primordial capillary plexus, indicating intussusceptive angiogenesis (Madureira and Soares, 2021). Angiopoietins are involved in the cross-talk between endothelial cells and pericytes (Madureira and Soares, 2021). The secretion of Ang-2 by endothelial cells is stimulated by hypoxic conditions as well as TNF-α, thrombin, and turbulent blood flow (Madureira and
留言 (0)