
記住我
CD3 bsAbs bind distinct antigens with each Fab arm, allowing them to simultaneously engage CD3 on a T cell and a tumor-associated antigen (TAA) expressed on tumor cells (1). CD3 bsAb-mediated cross-linking of these two cell types facilitates immunological synapse formation and subsequent T-cell activation, potentiating tumor cell kill via the secretion of cytolytic components and inflammatory cytokines (2, 3). By engaging CD3, bsAbs can recruit all available T cells to mediate tumor kill, regardless of their cognate specificity. As of December 2024, there are seven CD3 bsAbs with US Food and Drug Administration (FDA) approval for treatment of hematological cancers (Table 1) (4–7, 9–11). To date, the success in solid tumors remains limited, with two FDA approvals for Tebentafusp® (advanced uveal melanoma) (12) and Tarlatamab® (extensive-stage small cell lung cancer) (13). Despite these advances, there are still several hurdles for CD3 bsAb therapy in solid tumors (14) (Figure 1). Choosing a suitable tumor surface antigen is critical for clinical success, with consequences for an optimal therapeutic window and acceptable collateral damage to normal tissues. Additional hurdles include limited numbers of intratumoral T cells, an immunosuppressive tumor microenvironment (TME), and poor memory T-cell induction. We recently investigated several combination approaches to address these hurdles (15, 16), which are highlighted here within.
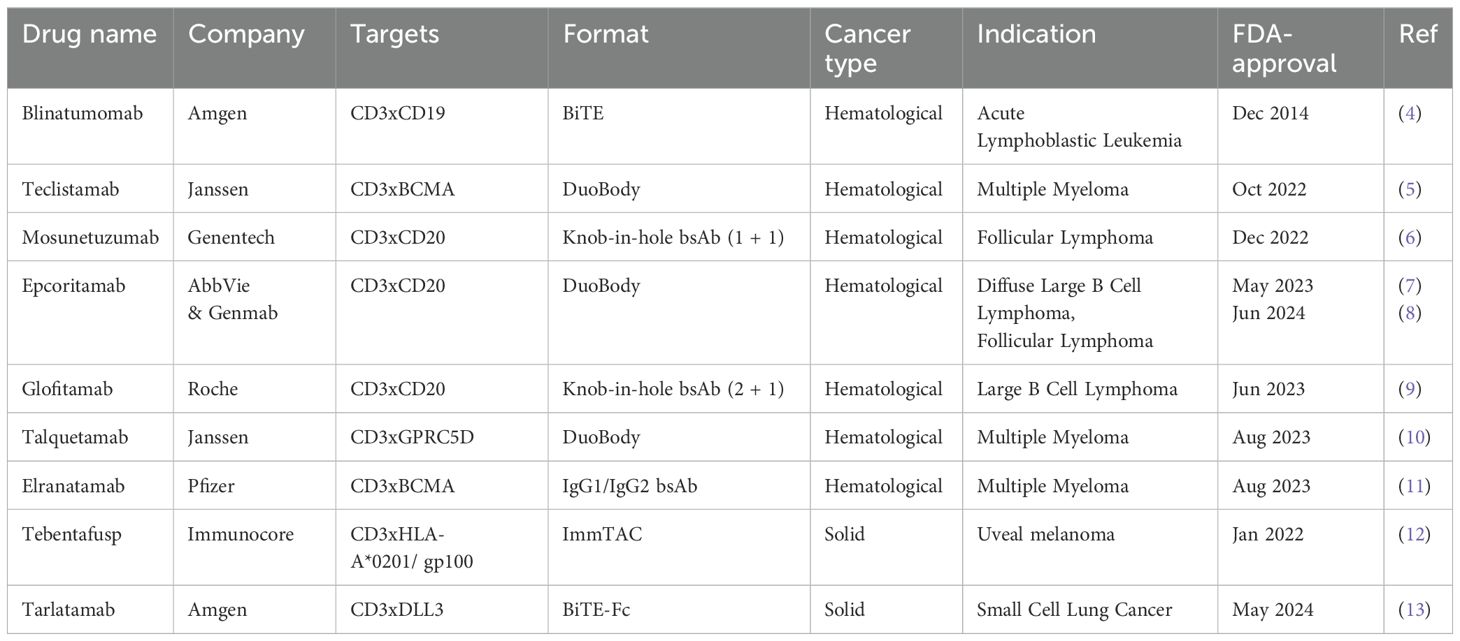
Table 1. FDA-approved CD3 bispecific therapies.
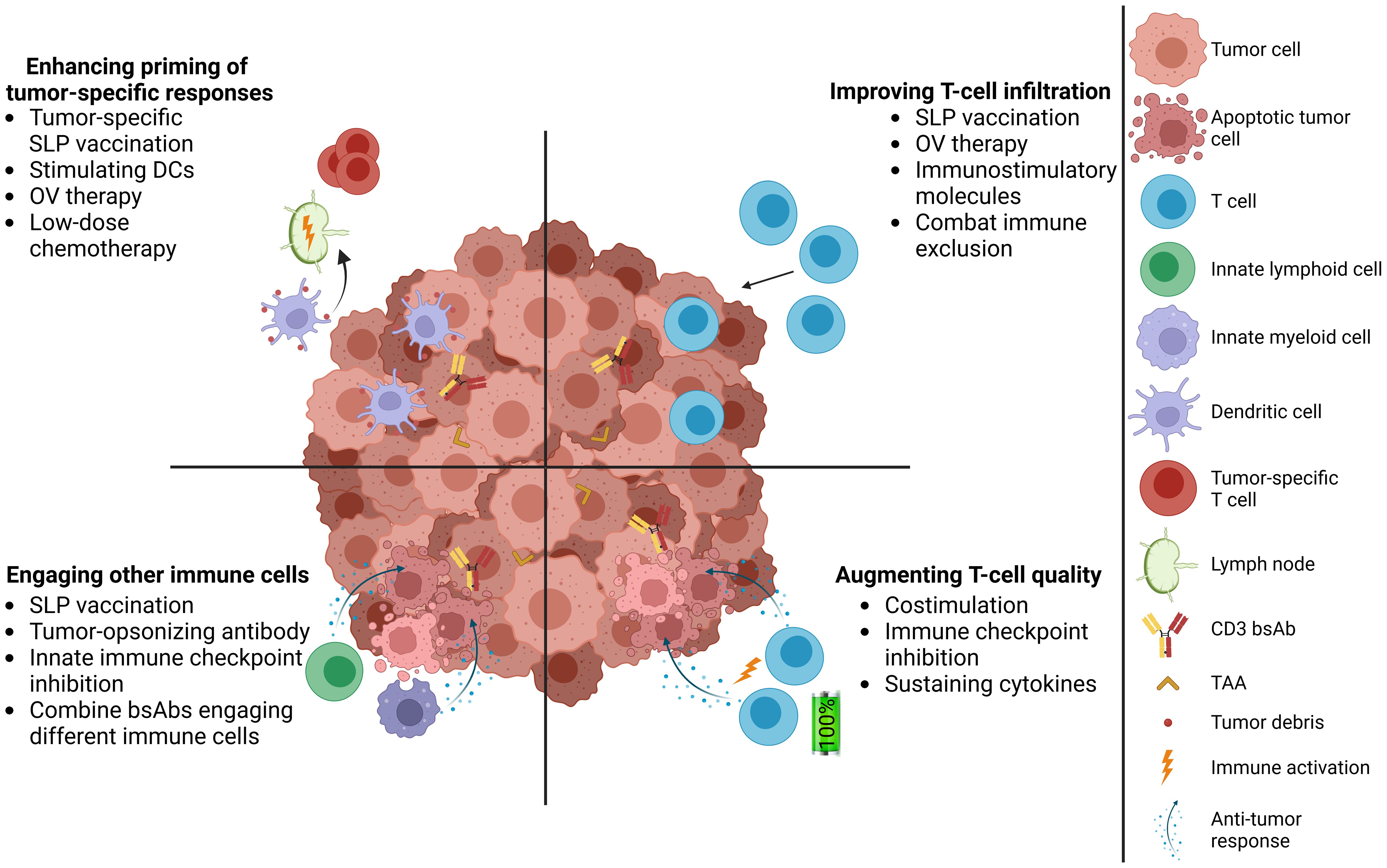
Figure 1. Complementing CD3 bsAb therapy with cancer vaccines in solid tumors. Addressing multiple hurdles of CD3 bsAb therapies, including promoting T-cell infiltration, increasing the quality of intratumoral T cells, engaging innate immune cells in the TME, and induction of tumor-specific T-cell responses. TAA, tumor-associated antigen; SLP, synthetic long peptide; DC, dendritic cell; OV, oncolytic virus.
Improving T-cell infiltration in solid tumorsIn contrast to hematological malignancies that are surrounded by T cells, solid cancers often harbor TMEs that contain limited T-cell numbers. This feature might be associated with impaired CD3 bsAb therapeutic efficacy in preclinical models (17), which is most prominent in immunologically “cold” tumors exhibiting poor T-cell infiltration (18). We recently showed in CXCR3 knock-out mice that CD3 bsAb antitumor activity in an immunologically “cold” tumor model was dependent on the influx of T cells (15, 19–22), supporting comparable preclinical findings that higher intratumoral T-cell numbers, either present at baseline or through therapy-induced influx, improve CD3 bsAb outcomes (20, 23).
We thus examined strategies that would boost intratumoral T-cell frequencies aiming to improve CD3 bsAb efficacy in immunology “cold” tumors. We reached significantly enhanced tumor eradication when T-cell responses were mounted via tumor nonspecific vaccines prior to CD3 bsAb (15). Vaccine adjuvants IL-2 and TLR7 agonist imiquimod, or TLR9 agonist CpG, were used to ensure sufficient co-stimulation in the lymphatics, enhancing T-cell priming and functionality. We observed that vaccination induced homing of activated CD8+ T cells towards the tumor rim and that subsequent CD3 bsAb administration further transformed T cells into potent effectors and enabled their deep infiltration into the tumor core (15). This process shares similarities with the proposed two-stage tumor-specific CD8+ T-cell activation previously reported (24), whereby initial activation in tumor-draining lymph nodes is followed by subsequent cytotoxic effector program acquisition after target recognition in the TME. We showed that tumor-specific or -nonspecific vaccination in combination with CD3 bsAb promoted a broadly inflamed TME, delayed tumor outgrowth and improved survival in mice (15). The irrelevance of vaccine antigen specificity to primary survival benefit is likely explained by unbiased trafficking of vaccine-activated T cells to non-lymphoid tissues, including the tumor, where engagement by the CD3 bsAb occurs regardless of the cognate specificity. Importantly, CD3 bsAb therapy with adjuvants alone did not improve tumor control in “cold” tumors, highlighting the necessity of an immunogenic antigen to generate substantial CD8 T-cell expansion and influx, resulting in significant survival benefits. Interestingly, combining chimeric antigen receptor (CAR) T-cell therapies with cognate antigen vaccination have shown superior engraftment, polyfunctionality and antitumor activity compared to non-vaccinated controls (25–27). Thus, vaccination could offer a relatively cheap “off-the-shelf” approach to systemically boost T cells during T-cell targeting therapies.
An alternative approach based on the same principle utilized oncolytic viruses (OV) to enhance CD3 bsAb therapy. Selective viral replication in tumor cells was shown to initiate a localized anti-viral immune response and strong influx of OV-specific T cells that could be subsequently engaged by CD3 bsAb (28). Others have reported similar survival benefits using intratumorally-injected OV constructs encoding for CD3 bsAbs (29), which limits antibody production to the tumor and potentially reduces systemic exposure and toxicities. However, this could result in CD3 bsAb not reaching distal tumors, whereas induction of an anti-viral response against the OV may either terminate bsAb production or, conversely, result in antigen and DAMP release. If both strategies demonstrate comparable clinical efficacy, the vaccination approach may be preferred to OVs, as it would not be limited to easily accessible tumors or by an anti-viral immune response, thereby allowing subsequent booster vaccinations to maintain T-cell influx.
Improving T-cell functionality in solid tumorsSolid cancers typically feature hostile and immunosuppressive TME, due to factors such as low pH, hypoxia, nutrient deprivation and high concentration of oxygen radicals, which can hamper cytotoxic T-cell effector functions (14). In addition, chronic exposure to antigens leads to exhaustion of T cells and expression of a plethora of inhibitory immune receptors. In the clinic, T-cell functionality also declines with prior rounds of systemic chemotherapy and disease progression, which may pose a challenge for CD3 bsAbs in heavily pre-treated patient populations (30, 31). There is some clinical data that correlate good T-cell functionality with clinical response to CD3 bsAb therapy in solid tumors, suggesting this is a likely contributing factor (32).
As immune checkpoint expression can be induced following CD3 bsAb therapy (33, 34), immune checkpoint inhibitors (ICIs) are frequently tested in combination with CD3 bsAbs, with many ongoing clinical trials for solid tumors (35), aimed at lifting the checkpoint-mediated brake on T-cell activation (Figure 1). Multiple preclinical studies show augmented T-cell functionality and therapeutic benefit when combing ICIs and CD3 bsAbs compared to CD3 bsAb monotherapy (36–38). Similarly, there are also indications that combining CAR T-cell therapy with ICIs could improve therapeutic outcomes (39). As we observed enhanced PD-1, NKG2A, CTLA-4, Tim-3 and TIGIT expression on T cells in mice treated with CD3 bsAb and vaccine combinations, suggesting that peripherally-derived T cells are more fit than TME-localized T cells (15), it would be interesting to test if the addition of ICIs could further enhance therapeutic efficacy.
Another strategy for improving T-cell functionality involves combining CD3 bsAbs with agonistic antibodies that trigger costimulatory signals. The concept is supported by second-generation CAR T-cell therapies which feature a costimulatory signaling domain, to provide a “signal 2” to T cells in addition to CD3 signaling (signal 1), resulting in superior T-cell persistence and functionality (40, 41). Preclinical studies have reported therapeutic benefit when combining CD3 bsAbs with agonistic antibodies providing costimulation via either 4-1BB or CD28 (42–44). Likewise, we observed improved tumor control when combining tumor-localized 4-1BB costimulation with CD3 bsAb therapy in mice inoculated with immunologically “cold” tumors (45). Given the boost in T-cell function and upregulation of immune checkpoints after T-cell activation, strategies combining CD3 bsAb with costimulatory signals seem promising for improving therapeutic benefit. However, further research is needed to compare the safety and efficacy of such combinations.
Tumor-specific responses are crucial for protective memory following CD3 bsAb therapyCD3 bsAb therapy has displayed impressive response rates in hematological malignancies in the clinic, but relapses are frequently reported (46–48). Tumor heterogeneity or downmodulation of surface expression of the targeted antigen is responsible for a large proportion of relapses, both for CD3 bsAbs and CAR T cells (46–50). While this phenomenon has not yet been described in the clinic for solid tumors, there are similar indications in preclinical studies (28), including ours, where escape of TRP1- cells was observed in end-stage tumors from CD3 bsAb-treated mice (45). To overcome this and decrease immune escape, combinations of multiple CD3 bsAbs or CAR T-cell specificities targeting different tumor antigens are being evaluated (51, 52).
An alternative approach to prevent escape through antigen loss is the induction of endogenous responses against antigen-negative tumor cells (i.e. antigen spread), which may arise after tumor antigen release upon tumor lysis and subsequent presentation by antigen presenting cells (APCs) (53). We previously reported that although CD3 bsAb monotherapy induced tumor-specific T-cell responses, these responses were short-lived and did not install protective memory in the immunologically “cold” B16F10 model (54). We also observed improved primary responses but no protection from secondary B16F10 tumor challenge when combining CD3 bsAb with 4-1BB costimulation, Fc-active tumor-opsonizing antibodies, or tumor-nonspecific vaccines (45).
Combining CD3 bsAb and tumor-specific vaccination enhanced survival against primary tumors and delayed tumor outgrowth upon secondary tumor challenge (45). However, complete protection against tumor rechallenge was not reached, implying there was insufficient antigen spread in the immunologically “cold” tumor model. Therefore, we hypothesized that the expression of a highly immunogenic antigen may be required for functional memory. In the immunologically “hot” MC38 tumor model, tumor antigen spreading occurred independent of the administered treatment. Tumor-specific T cells were detected in all mice, and most mice that had completely eradicated the first tumor were protected from MC38 tumor rechallenge (45). Protection against rechallenge was completely ablated upon CD8+ T-cell depletion, highlighting the necessity of tumor-specific endogenous CD8+ T-cell responses for long-term memory. There was substantial outgrowth of antigen-negative tumor cells in mice treated with CD3 bsAb monotherapy, but not when CD3 bsAb was combined with vaccination, highlighting the need to strike hard during primary tumor treatment to prevent early immune escape (45).
Our results suggest that “off-the-shelf” T-cell stimulating vaccines can be utilized to enhance CD3 bsAb anti-tumor activity against primary solid tumors. However, preventing relapse requires a level of intrinsic tumor immunogenicity and can be further bolstered by tumor-specific immunization (Figure 1). Such combinations could shortly become a reality given the recent success of personalized mRNA cancer vaccines in the clinic (55). Given the local and potent immune activation and concomitant enhanced T-cell infiltration with OVs, these could also serve as an alternative CD3 bsAb combination partner to drive endogenous anti-tumor T-cell responses (56).
In summary, CD3 bsAbs and vaccine combinations improved survival against primary tumors and tumor rechallenge in mice. The immune status of tumors impacted survival benefit, where CD3 bsAb anti-tumor activity in tested immunologically “hot” tumor models benefited from innate activators, whereas “cold” tumor models required provision of an immunogenic tumor antigen. It remains to be determined if combinations with novel therapies such as personalized mRNA vaccines can benefit CD3 bsAb therapy in the clinic.
Improving CD3 bsAb therapy through engagement of other immune cellsEmerging evidence suggests that a combination of both innate and adaptive immunity is pivotal for complete and durable tumor eradication following immunotherapy (57–60) and understanding immune cell interplay in the TME can thus further improve therapeutic efficacy. Recently, we reported that the CD3 bsAb and vaccine combination increased frequencies of intratumoral CD8+ T cells, NK cells, inflammatory macrophages and neutrophils in the B16F10 model (16), supporting previous findings that CD3 bsAb therapy enhances NK-cell and macrophage activation (61). We revealed that CD3 bsAb-activated T cells attracted macrophages into the tumor and skewed them towards a late-stage, pro-inflammatory phenotype (16). These macrophages were crucial for the anti-tumor activity of the CD3 bsAb and vaccination combination, highlighting the importance of a coordinated innate and adaptive immune response for effective tumor control (16). Furthermore, comparable late-stage pro-inflammatory macrophage subsets were identified in human cancers by gene expression analysis (62), which were found to correlate with ICI treatment response in a breast cancer cohort (16, 63). Our findings align with studies demonstrating crucial roles for innate effector cells in immunotherapy (16, 60, 64), supporting the rationale for the so far unexplored combination of innate cell-stimulating therapeutics with CD3 bsAbs. There is a number of novel therapeutic strategies seeking to empower innate responses against cancer, including stimulation of pattern recognition receptors like stimulator of interferon genes (GAS-STING), TLR, and RIG-I-like receptors (RLR), as well as modulation of macrophages and natural killer cells (65). Immune-stimulating antibody conjugates (ISAC) combine therapeutic antibodies and innate-stimulating payloads, such as TLR or STING agonists, to promote inflammatory TMEs (66). Several ISACs have been clinically evaluated, including Bolt Biotherapeutic’s anti-HER2-TLR7/8 (BDC-1001) for HER2-positive solid tumors (NCT04278144) and Takeda’s anti-CCR2-STING (TAK-500) for various solid tumors (NCT05070247). Another way to potentially improve intratumoral T-cell functionality and boost innate effectors during CD3 bsAb therapy is to co-administer sustaining cytokines, similar to strategies using genetically engineered CAR T cells that express cytokines upon activation for improved therapeutic outcomes (67). There are reports showing positive contributions of cytokines to CD3 bsAb therapy preclinically, including combinations with immunomodulating drugs that induce IL-2 (68), Fc-fused IL-15 compounds (69), and bsAb-IL-15 fusions (70). Although the clinical efficacy of innate activators remains to be elucidated, especially in relation to combinations with CD3 bsAb, the recent FDA approval of ImmunityBio’s IL-15 receptor agonist with Bacillus Calmette-Guérin (BCG) vaccination for bladder cancer highlights the potential of next-generation innate stimulators for cancer immunotherapy (71).
Harnessing innate immune cells could greatly potentiate CD3 bsAb efficacy. As one of the safety risks of CD3 bsAb therapy is cytokine release syndrome, it remains to be determined if this is exacerbated by combinations with vaccination and/or innate stimulators. Furthermore, it will be important to consider that innate immune cell engagement could be context-dependent, as engagement of predominantly immunosuppressive immune cells might even lower therapeutic outcomes.
Remaining questions and path to clinical applicationTo ensure clinical success of CD3 bsAb and T-cell stimulating vaccine combination therapies, numerous unanswered questions and knowledge gaps must be addressed. For example, deeper understanding on vaccine prerequisites for sufficient T-cell priming prior to CD3 bsAb therapy is needed. Ideally, vaccines should elicit a Th1 response in CD8+ T cells. Most marketed childhood and travelers’ vaccines are prophylactic in nature, designed to induce neutralizing Abs, requiring (pre)clinical studies to determine their suitability for inducing Th1 responses supporting CD8+ cytolytic T cells. In addition to their wide availability, a key advantage of approved prophylactic vaccines is their well-established safety profiles, facilitating clinical evaluation with an experimental CD3 bsAb agent. T-cell recall responses from previous immunizations could be utilized (Supplementary Table), as retrospective analyses suggest beneficial associations between ICI outcomes and earlier Covid-19 or influenza vaccination (72–74). However, prospective randomized clinical trials are required to fully understand if prophylactic tumor non-specific vaccinations can improve immunotherapy efficacy in human cancers. Our murine studies suggest that tumor-specific vaccination may be a more suitable choice, owing to its ability to improve both survival against primary tumor and protection against tumor recurrence (45). Cancer vaccine development has historically faced challenges with limited success (75–77). Recent advances in the field of precision medicine and mRNA-technologies may help overcome the shortcomings of traditional cancer vaccines and potentially revolutionize cancer immunotherapy. Many next-generation cancer vaccines are in clinical trials in combination with ICI (55, 78, 79), and given our preclinical results, it would be of interest to explore their efficacy in combination with CD3 bsAbs.
Further understanding of the role of adjuvants is warranted, as this may affect clinical applicability of the approach. Since we depicted a pivotal role for T-cell and macrophage cooperation in therapeutic response (16), it may be interesting to evaluate which vaccine adjuvants or innate activators are optimal to prime innate cells for contributing to CD3 bsAb efficacy. Recent phase II trial data for patients with HPV-induced abnormal cervical cells showed localized treatment with imiquimod promoted regression of high-grade lesions and significantly increased T-cell infiltration in the cervix (80). The addition of an HPV vaccine had no impact on therapeutic outcome (80), similar to our preclinical observations that tumor-specific vaccination with adjuvants imiquimod and IL-2 was insufficient for tumor control (15). Since CD3 bsAb and tumor-specific vaccine combinations enhanced tumor control in our study (15), combining CD3 bsAb with imiquimod could further improve treatment outcomes for cervical indications.
Another important area of research is the impact of baseline T-cell fitness on treatment outcome. The phenotype and functional status of endogenous circulating T cells at baseline are emerging as important parameters for the clinical activity for T-cell engagers (17, 32, 81, 82). The requirement of baseline T-cell infiltration in tumor nests for therapeutic efficacy is unclear, as shown recently for CD3xCEA in MSS-CRC patients (83). In addition, the two FDA-approved CD3 bsAbs for solid tumors show efficacy in indications that are notorious for being immunologically “cold” and for which ICI therapy is largely ineffective (12, 13). These findings suggest the initial T-cell activation signal may come from a limited number of T cells interacting with tumor cells at the edge of the tumor nests, resulting in a gradient of chemokines summoning circulating T cells to tumor sites, thus promoting T-cell infiltration (83). Since we showed that combining CD3 bsAb with vaccination helped recruit fresh peripheral T cells to the tumor via the CXCR3 axis in mouse models, clinical studies are warranted to evaluate if influx of peripheral cells into the tumor also occurs in humans and how priming may impact T-cell fitness pre- and post-CD3 bsAb administration.
Finally, successful clinical translation will need to enhance CD3 bsAb efficacy whilst maintaining an acceptable safety window. Activation of T cells by vaccination followed by CD3 bsAb, with or without additional innate activators, could drive immune-related adverse events. Furthermore, we need to understand vaccine response kinetics in relation to CD3 bsAb administration and whether there is a preference for either recall or de novo response. Rational bsAb design, careful selection of the target antigen, and dose-optimization strategies could mitigate potential dose-limiting toxicities, as well as potentially reinforcing clinical activity of CD3 bsAbs in solid tumors (83–86) (Figure 1).
To conclude, we advocate for the clinical translation of T-cell stimulating vaccines during CD3 bsAb therapy for superior efficacy against primary tumors and to potentially drive immunological memory. The combination could offer a versatile and effective means to improve cancer immunotherapy outcomes, which may be applicable to various tumor types, immunotherapies, and vaccine modalities.
Author contributionsKL: Conceptualization, Writing – review & editing, Writing – original draft. JM: Validation, Writing – review & editing. VO: Conceptualization, Writing – original draft, Writing – review & editing. NP: Writing – original draft, Writing – review & editing. KK: Conceptualization, Supervision, Writing – review & editing. Tv: Conceptualization, Supervision, Writing – review & editing.
FundingThe author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interestAuthors KL, VO, NP, and KK were employed by the company Genmab B.V.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1548446/full#supplementary-material
References1. Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery. (2019) 18:585–608. doi: 10.1038/s41573-019-0028-1
PubMed Abstract | Crossref Full Text | Google Scholar
2. Roda-Navarro P, Álvarez-Vallina L. Understanding the spatial topology of artificial immunological synapses assembled in T cell-redirecting strategies: A major issue in cancer immunotherapy. Front Cell Dev Biol. (2020) 7:370. doi: 10.3389/fcell.2019.00370
PubMed Abstract | Crossref Full Text | Google Scholar
4. Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. (2017) 376:836–47. doi: 10.1056/NEJMoa1609783
PubMed Abstract | Crossref Full Text | Google Scholar
5. Moreau P, Garfall AL, van de Donk N, Nahi H, San-Miguel JF, Oriol A, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. (2022) 387:495–505. doi: 10.1056/NEJMoa2203478
PubMed Abstract | Crossref Full Text | Google Scholar
6. Budde LE, Sehn LH, Matasar M, Schuster SJ, Assouline S, Giri P, et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol. (2022) 23:1055–65. doi: 10.1016/S1470-2045(22)00335-7
PubMed Abstract | Crossref Full Text | Google Scholar
7. Thieblemont C, Phillips T, Ghesquieres H, Cheah CY, Clausen MR, Cunningham D, et al. Epcoritamab, a novel, subcutaneous CD3xCD20 bispecific T-cell-engaging antibody, in relapsed or refractory large B-cell lymphoma: dose expansion in a phase I/II trial. J Clin Oncol. (2023) 41:2238–47. doi: 10.1200/JCO.22.01725
PubMed Abstract | Crossref Full Text | Google Scholar
8. Vose J, Vitolo U, Lugtenburg P, Chamuleau MED, Linton KM, Thieblemont C, et al. EPCORE NHL-1 follicular lymphoma (FL) cycle (C) 1 optimization (OPT) cohort: Expanding the clinical utility of epcoritamab in relapsed or refractory (R/R) FL. J Clin Oncol. (2024) 42:7015. doi: 10.1200/JCO.2024.42.16_suppl.7015
Crossref Full Text | Google Scholar
9. Dickinson MJ, Carlo-Stella C, Morschhauser F, Bachy E, Corradini P, Iacoboni G, et al. Glofitamab for relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2022) 387:2220–31. doi: 10.1056/NEJMoa2206913
PubMed Abstract | Crossref Full Text | Google Scholar
10. Chari A, Minnema MC, Berdeja JG, Oriol A, van de Donk N, Rodríguez-Otero P, et al. Talquetamab, a T-cell-redirecting GPRC5D bispecific antibody for multiple myeloma. N Engl J Med. (2022) 387:2232–44. doi: 10.1056/NEJMoa2204591
PubMed Abstract | Crossref Full Text | Google Scholar
11. Lesokhin AM, Tomasson MH, Arnulf B, Bahlis NJ, Miles Prince H, Niesvizky R, et al. Elranatamab in relapsed or refractory multiple myeloma: phase 2 MagnetisMM-3 trial results. Nat Med. (2023) 29:2259–67. doi: 10.1038/s41591-023-02528-9
PubMed Abstract | Crossref Full Text | Google Scholar
12. Nathan P, Hassel JC, Rutkowski P, Baurain JF, Butler MO, Schlaak M, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med. (2021) 385:1196–206. doi: 10.1056/NEJMoa2103485
PubMed Abstract | Crossref Full Text | Google Scholar
13. Ahn MJ, Cho BC, Felip E, Korantzis I, Ohashi K, Majem M, et al. Tarlatamab for patients with previously treated small-cell lung cancer. N Engl J Med. (2023) 389:2063–75. doi: 10.1056/NEJMoa2307980
PubMed Abstract | Crossref Full Text | Google Scholar
14. Middelburg J, Kemper K, Engelberts P, Labrijn AF, Schuurman J, van Hall T. Overcoming challenges for CD3-bispecific antibody therapy in solid tumors. Cancers (Basel). (2021) 13:287. doi: 10.3390/cancers13020287
PubMed Abstract | Crossref Full Text | Google Scholar
15. Middelburg J, Sluijter M, Schaap G, Göynük B, Lloyd K, Ovcinnikovs V, et al. T-cell stimulating vaccines empower CD3 bispecific antibody therapy in solid tumors. Nat Commun. (2024) 15:48. doi: 10.1038/s41467-023-44308-6
PubMed Abstract | Crossref Full Text | Google Scholar
16. van Elsas MJ, Middelburg J, Labrie C, Roelands J, Schaap G, Sluijter M, et al. Immunotherapy-activated T cells recruit and skew late-stage activated M1-like macrophages that are critical for therapeutic efficacy. Cancer Cell. (2024) 42:1032–50.e10. doi: 10.1016/j.ccell.2024.04.011
PubMed Abstract | Crossref Full Text | Google Scholar
17. Ströhlein MA, Lefering R, Bulian DR, Heiss MM. Relative lymphocyte count is a prognostic parameter in cancer patients with catumaxomab immunotherapy. Med Hypotheses. (2014) 82:295–9. doi: 10.1016/j.mehy.2013.12.014
PubMed Abstract | Crossref Full Text | Google Scholar
18. Patel D, Balderes P, Lahiji A, Melchior M, Ng S, Bassi R, et al. Generation and characterization of a therapeutic human antibody to melanoma antigen TYRP1. Hum Antibodies. (2007) 16:127–36. doi: 10.3233/HAB-2007-163-407
PubMed Abstract | Crossref Full Text | Google Scholar
19. Belmontes B, Sawant DV, Zhong W, Tan H, Kaul A, Aeffner F, et al. Immunotherapy combinations overcome resistance to bispecific T cell engager treatment in T cell-cold solid tumors. Sci Transl Med. (2021) 13:eabd1524. doi: 10.1126/scitranslmed.abd1524
PubMed Abstract | Crossref Full Text | Google Scholar
20. Cremasco F, Menietti E, Speziale D, Sam J, Sammicheli S, Richard M, et al. Cross-linking of T cell to B cell lymphoma by the T cell bispecific antibody CD20-TCB induces IFNγ/CXCL10-dependent peripheral T cell recruitment in humanized murine model. PloS One. (2021) 16:e0241091. doi: 10.1371/journal.pone.0241091
PubMed Abstract | Crossref Full Text | Google Scholar
21. Li J, Ybarra R, Mak J, Herault A, De Almeida P, Arrazate A, et al. IFNgamma-induced chemokines are required for CXCR3-mediated T-cell recruitment and antitumor efficacy of anti-HER2/CD3 bispecific antibody. Clin Cancer Res. (2018) 24:6447–58. doi: 10.1158/1078-0432.CCR-18-1139
PubMed Abstract | Crossref Full Text | Google Scholar
22. You R, Artichoker J, Ray A, Gonzalez Velozo H, Rock DA, Conner KP, et al. Visualizing spatial and stoichiometric barriers to bispecific T-cell engager efficacy. Cancer Immunol Res. (2022) 10:698–712. doi: 10.1158/2326-6066.CIR-21-0594
PubMed Abstract | Crossref Full Text | Google Scholar
23. Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-fcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. (2017) 31:383–95. doi: 10.1016/j.ccell.2017.02.001
PubMed Abstract | Crossref Full Text | Google Scholar
24. Prokhnevska N, Cardenas MA, Valanparambil RM, Sobierajska E, Barwick BG, Jansen C, et al. CD8(+) T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity. (2023) 56:107–24.e5. doi: 10.1016/j.immuni.2022.12.002
PubMed Abstract | Crossref Full Text | Google Scholar
25. Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. (2019) 365:162–8. doi: 10.1126/science.aav8692
PubMed Abstract | Crossref Full Text | Google Scholar
26. Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. (2020) 367:446–53. doi: 10.1126/science.aay5967
PubMed Abstract | Crossref Full Text | Google Scholar
27. Wang X, Diamond DJ, Forman SJ, Nakamura R. Development of CMV-CD19 bi-specific CAR T cells with post-infusion in vivo boost using an anti-CMV vaccine. Int J Hematol. (2021) 114:544–53. doi: 10.1007/s12185-021-03215-6
PubMed Abstract | Crossref Full Text | Google Scholar
28. Groeneveldt C, Kinderman P, van den Wollenberg DJM, van den Oever RL, Middelburg J, Mustafa DAM, et al. Preconditioning of the tumor microenvironment with oncolytic reovirus converts CD3-bispecific antibody treatment into effective immunotherapy. J Immunother Cancer. (2020) 8:e001191. doi: 10.1136/jitc-2020-001191
PubMed Abstract | Crossref Full Text | Google Scholar
29. Wang Q, Ma X, Wu H, Zhao C, Chen J, Li R, et al. Oncolytic adenovirus with MUC16-BiTE shows enhanced antitumor immune response by reversing the tumor microenvironment in PDX model of ovarian cancer. Oncoimmunology. (2022) 11:2096362. doi: 10.1080/2162402X.2022.2096362
PubMed Abstract | Crossref Full Text | Google Scholar
30. Petersen CT, Hassan M, Morris AB, Jeffery J, Lee K, Jagirdar N, et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kdelta inhibitors and VIP antagonists. Blood Adv. (2018) 2:210–23. doi: 10.1182/bloodadvances.2017011254
PubMed Abstract | Crossref Full Text | Google Scholar
31. Verma R, Foster RE, Horgan K, Mounsey K, Nixon H, Smalle N, et al. Lymphocyte depletion and repopulation after chemotherapy for primary breast cancer. Breast Cancer Res. (2016) 18:10. doi: 10.1186/s13058-015-0669-x
PubMed Abstract | Crossref Full Text | Google Scholar
32. Cortes-Selva D, Perova T, Skerget S, Vishwamitra D, Stein S, Boominathan R, et al. Correlation of immune fitness with response to teclistamab in relapsed/refractory multiple myeloma in the MajesTEC-1 study. Blood. (2024) 144:615–28. doi: 10.1182/blood.2023022823
PubMed Abstract | Crossref Full Text | Google Scholar
33. Hipp S, Voynov V, Drobits-Handl B, Giragossian C, Trapani F, Nixon AE, et al. A bispecific DLL3/CD3 igG-like T-cell engaging antibody induces antitumor responses in small cell lung cancer. Clin Cancer Res. (2020) 26:5258–68. doi: 10.1158/1078-0432.CCR-20-0926
PubMed Abstract | Crossref Full Text | Google Scholar
34. Kohnke T, Krupka C, Tischer J, Knosel T, Subklewe M. Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J Hematol Oncol. (2015) 8:111. doi: 10.1186/s13045-015-0213-6
PubMed Abstract | Crossref Full Text | Google Scholar
35. Zhu WM, Middleton MR. Combination therapies for the optimisation of Bispecific T-cell Engagers in cancer treatment. Immunother Adv. (2023) 3:ltad013. doi: 10.1093/immadv/ltad013
PubMed Abstract | Crossref Full Text | Google Scholar
36. Chang CH, Wang Y, Li R, Rossi DL, Liu D, Rossi EA, et al. Combination therapy with bispecific antibodies and PD-1 blockade enhances the antitumor potency of T cells. Cancer Res. (2017) 77:5384–94. doi: 10.1158/0008-5472.CAN-16-3431
PubMed Abstract | Crossref Full Text | Google Scholar
37. Crawford A, Haber L, Kelly MP, Vazzana K, Canova L, Ram P, et al. A Mucin 16 bispecific T cell-engaging antibody for the treatment of ovarian cancer. Sci Transl Med. (2019) 11:e7534. doi: 10.1126/scitranslmed.aau7534
留言 (0)