
記住我
Sugarcane holds significant importance as a global crop, serving as a vital source of sugar and biofuel for millions of people. However, its production faces a serious and widespread threat from disease, with leaf scald (LS) being one of the most important. Caused by the xylem-inhabiting bacterium Xanthomonas albilineans (Ashby) Dowson (Xalb) (Garces et al., 2014), LS has been reported in over 60 countries. Particularly affected are Australia, United States, Philippines, Vietnam, and Thailand (Rott and Davis, 2000). The origins of LS in Australia can be traced back to 1911, with subsequent spread across all sugarcane-growing regions (Rott and Davis, 1995; Davis et al., 1997). The LS disease impact may be severe, leading to significant yield losses, reduced juice quality, and in some cases, the complete loss of entire sugarcane fields (Ricaud and Ryan, 1989; Rott and Davis, 2000; Ntambo et al., 2019). The consequences of LS’s presence are so pronounced that approximately 20% of potentially high-yielding sugarcane cultivars in Australia are rejected due to their susceptibility to this disease (Birch, 2001). This alarming situation has prompted the recognition of LS as a top-priority pest threat in the national plant biosecurity status report. As researchers and authorities work to find solutions, preserving the health and productivity of sugarcane crops remains of utmost importance (Plant Health Australia, 2019).
Xalb infects the xylem vessels of sugarcane plants, giving rise to distinctive white-yellow pencil lines along the primary leaf veins. In severe cases, this bacterial infection leads to chlorosis, necrosis, and even the death of the entire plant (Rott and Davis, 2000; Lin et al., 2018). The pathogen is known to produce a potent pathotoxin called albicidin, which acts as a DNA gyrase inhibitor, causing foliar symptoms by hindering the replication of chloroplast DNA (Birch and Patil, 1985). Additionally, albicidin is believed to facilitate systemic invasion and potentially trigger the transition from a latent to an active disease state (Cociancich et al., 2015). Interestingly, many sugarcane cultivars can tolerate the presence of the pathogen without displaying any visible symptoms, or the symptoms may be so mild that they go unnoticed (Ricaud and Ryan, 1989; Rott and Davis, 1995; Rott et al., 1997). Unfortunately, this latent infection can lead to the inadvertent distribution of infected seed cane across the sugarcane production regions. Given these complexities, accurately detecting Xalb is crucial for effective LS diagnosis and management. Therefore, it is important to develop methodologies that offer high sensitivity and specificity in quantitative Xalb detection.
Traditionally, the diagnosis of LS disease and the assessment of sugarcane test cultivars for LS resistance have relied on observing phenotypic symptoms. However, the latent and erratic nature of symptom expression makes visual disease diagnosis challenging and can lead to the mislabeling of susceptible cultivars as “resistant.” This difficulty in accurately identifying infected plants has been a significant contributor to the global spread of LS through seemingly “healthy” planting materials (Rott, 1995). To address these challenges, various alternative approaches have been explored for LS diagnosis. These methods include isolation on selective media (Davis et al., 1994), microscopy (Mensi et al., 2014), ELISA (enzyme-linked immunosorbent assay) (Wang et al., 1999), PCR (polymerase chain reaction) (Dias et al., 2018), and qPCR (quantitative polymerase chain reaction) (Garces et al., 2014). However, each of these techniques has its limitations. Isolation on selective media and microscopy can be highly effective, especially for detecting Xalb in symptomless plants, but is a cumbersome and time-consuming process (Wang et al., 1999; Mensi et al., 2014). Immunological and molecular methods offer varying degrees of sensitivity for detecting Xalb, but their wide application has been limited due to the need for complex equipment and advanced laboratory facilities, and reliance on expensive commercial kits or complex chemical-based laborious and time-consuming DNA extraction/purification procedures (Pan et al., 1999; Garces et al., 2014). These factors present obstacles in developing an on-farm rapid Xalb detection method and underscore the need for a straightforward, rapid, and cost-effective Xalb detection method that minimizes or eliminates the need for chemical reagents in sample preparation.
In this study, we present a streamlined method for the rapid isolation and in-situ quantification of Xalb DNA. This approach leverages a direct heat lysis approach to release DNA from infected sugarcane samples, avoiding the need for costly commercial kits or complex reagents. This reagent-free DNA isolation method, combined with loop-mediated isothermal amplification (LAMP), offers a simple, rapid solution for Xalb detection and quantification. LAMP is known for its efficiency, low cost, and effectiveness in detecting plant pathogens even in crude extracts, making it more resilient than PCR-based methods, which may be affected by amplification inhibitors (Tsai et al., 2009; Francois et al., 2011). Additionally, the method provides both qualitative detection (visual or naked-eye assessment) and quantitative measurement of Xalb DNA.
Therefore, this study thus aimed to: (i) develop a simplified, LAMP-based diagnostic method paired with a rapid, reagent-free DNA isolation technique for in-situ Xalb detection and quantification from various sugarcane samples; (ii) validate the diagnostic’s quantitative accuracy through qPCR; (iii) assess for correlation of this two-stage diagnostic approach with previously established LS ratings across sugarcane cultivars with known resistance; and (iv) identify optimal sampling times and plant parts for application of the developed diagnostic by tracking disease progression over time.
2 Materials and methods 2.1 Reagents and materialsAll reagents and chemicals used were of analytical grade. Nuclease-free water from Integrated DNA Technologies, Australia, was employed for preparing all aqueous solutions. The PureLink™ Microbiome DNA purification kit was purchased from Thermo Fisher Scientific, Australia. Additionally, all designed primers and the synthetic target (4.2 × 107 copies/μL) were purchased from Integrated DNA Technologies, United States. The WarmStart® Colorimetric LAMP 2X Master Mix and WarmStart® Multi-Purpose RT-LAMP 2X Master Mix, as well as the 2X SensiFAST SYBER No-ROX Master Mix, were obtained from New England Biolabs, Ipswich, MA, United States, and Meridian Bioscience, Cincinnati, Ohio, United States, respectively. Furthermore, all other reagents were purchased from Sigma-Aldrich, United States.
2.2 Source of bacteria and culture conditionsThe Xalb strain 3/14/9 bacterium was obtained from the Sugar Research Australia (SRA), Indooroopilly Research Station, Indooroopilly, Queensland, Australia (S 27.50°, E 152.98°). It was then cultured in the modified liquid broth Wilbrink’s media at a temperature of 28°C for 5 days, following the protocol of Davis et al. (1994). Leifsonia xyli subsp. xyli (Lxx), the bacterium responsible for sugarcane ratoon stunting disease (RSD), was used as a negative control. Lxx was isolated from an infected sugarcane plant at the SRA Woodford Pathology Research Station (S 26.93°, E 152.78°) in Queensland, Australia. It was cultured in a modified liquid S8 medium and incubated at 28°C for 4 weeks with gentle shaking at 200 rpm in the dark, following the protocols of Davis et al. (1980) and Chakraborty et al. (2024).
2.3 Field trial establishment, inoculation, and plantingThree separate field trials with the same cultivars were conducted at the SRA Pathology Research Station in Woodford (S 26.93°, E 152.78°) for this study from 2022–2023. For this, 10 sugarcane cultivars were obtained from a disease-free propagation block at the same research facility, which was established using sugarcane setts treated with long hot water (50°C for 2 h) with a sterile cane knife to eliminate systemic Xalb infections (Table 1). Two-month-old plants were inoculated with Xalb 2–3 months before sampling using the protocol of Koike (1965). On a cloudy day, the cane tops of six plants per cultivar were decapitated using sharp short, handled cane knives above the growing point between the 3rd and 4th dewlap. Subsequently, the Xalb bacteria were “painted” onto the cut tissue with a brush dipped in the inoculum (~108 cells/mL). Each replicated plot comprises 3 m (length) × 1.5 m (width) with a 1 m gap between two plots. The soil preparation for the trial involved maintaining good soil structure (red and grey soils) and ensuring moisture conservation through minimum tillage practices. The region receives an annual rainfall of 1,200–1,400 mm, supporting optimal soil conditions for the experiment. The trials were established using a randomized complete block (RCB) design, with three replications, each consisting of six plants.
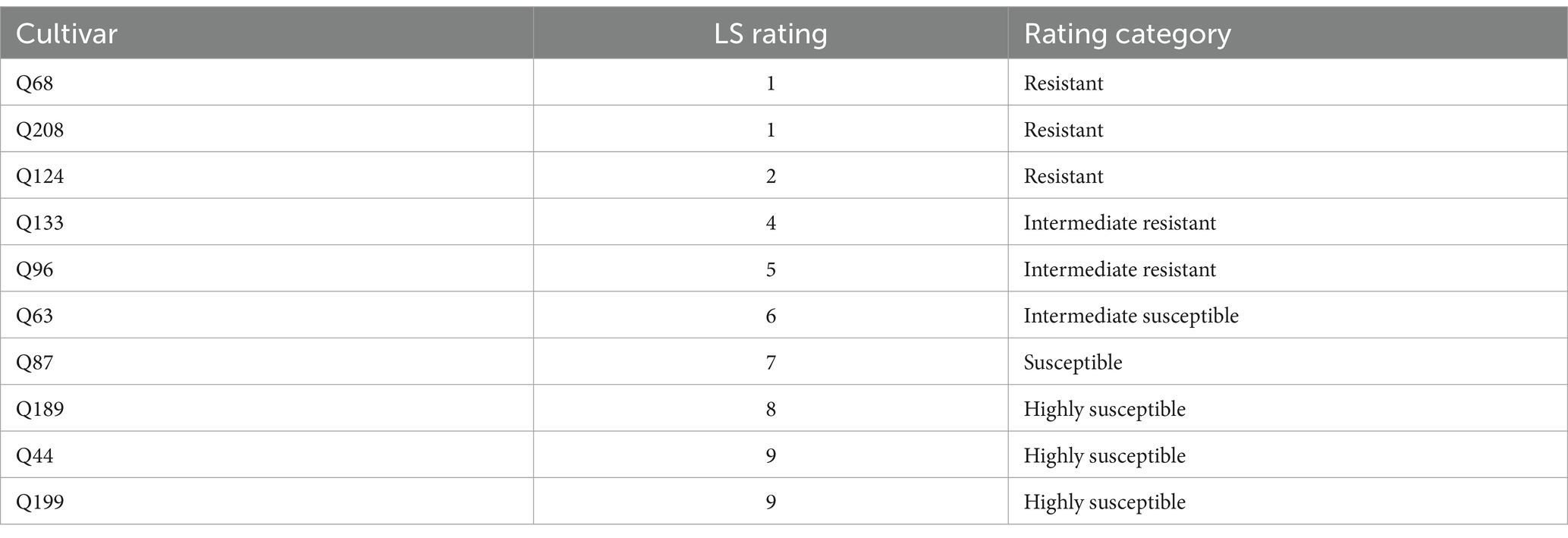
Table 1. List of sugarcane cultivars used and their LS disease ratings, categorized by phenotypic symptoms (Rott et al., 1997).
2.4 Sugarcane field sample collectionSugarcane leaf tissues, meristematic tissues, and xylem sap were sampled from three stalks (one stalk/replication of each cultivar) at different time points of post-inoculation from the SRA Woodford LS field trial in July 2022, October 2022, and January 2023 for the first trial. For xylem sap, vascular extracts (2 mL/cultivar) were obtained using positive air pressure extraction, as outlined by Croft et al. (1994). The extracts were kept on ice during transport to the laboratory and subsequently stored at −20°C until further processing. To validate the assay’s performance using field-derived samples, sugarcane samples were collected from eight cultivars (Q68, Q208, Q124, Q133, Q96, Q63, Q87, and Q44) with various disease ratings (Table 1) initially included in the first trial. In subsequent trials, two additional highly susceptible cultivars (Q189 and Q199) were included to obtain more consistent results (Table 1).
To determine the best leaf position for detecting Xalb presence, leaf tissues were collected from three locations on each of the three plants across all 10 sugarcane cultivars (Table 1). These positions were chosen based on their relevance to Xalb detection, ensuring consistent and reliable sampling. Leaf tissue samples were collected monthly from the second LS trial between May 2023 and December 2023 to observe consistency at different time points. Samples were taken from the top visible dewlap (TVD) leaf, the topmost fully expanded leaf with a visible dewlap, as well as from TVD +1 and TVD −1, the leaves immediately above and below the TVD leaf, respectively. These three positions served as standard reference points for consistent sampling. To track Xalb population development in the leaf tissues, xylem sap, and meristematic tissues of the highly susceptible (Q44) and resistant (Q208) cultivars over time, samples were collected monthly from October 2022 to October 2023 from the third LS field trial at SRA Woodford following the same protocols described above. LS disease resistance was assessed over multiple years based on visible symptoms for most of the cultivars (Rott et al., 1997), as detailed in Table 1.
2.5 DNA extractionGenomic DNA from cultured Xalb cells and LS-infected sugarcane samples was extracted for qPCR analysis using the protocol provided in The PureLink™ Microbiome DNA purification kit manual (Thermo Fisher Scientific, Australia). Each experiment was performed in triplicate with three replications to ensure consistency.
For the detection of the Xalb bacteria via LAMP assay, a rapid, reagent-free method for DNA isolation was utilized, as detailed by Chakraborty et al. (2024). The concentration of bacteria was estimated subsequently via comparison against a standard curve of known Xalb amounts and consequential LAMP detection limitation was determined. To establish the standard curve, a series of titrated cultured Xalb cells (ranging from 107 to 100 cells/μL) was introduced into disease-free sugarcane xylem sap samples (100 μL). These sap samples were collected from SRA’s disease-free propagation block, confirmed as Xalb-free by qPCR, and then subjected to boiling at 95°C for 2 min in a heat block. Subsequently, 2 μL of the boiled suspension was pipetted directly into LAMP or qPCR reactions. Each concentration was replicated three times.
Following the establishment of standard detection curves, field collected leaf tissue and meristematic tissue samples were cut into small pieces and incubated in 100 μL of distilled water for 10 min. The sample solution was then heat lysed by boiling at 95°C for 2 min in a heat block, and 2 μL of the suspension was used for further analysis. Similarly, for the xylem sap samples, 100 μL of sap was directly boiled at 95°C for 2 min without any incubation, and 2 μL of the suspension was used for analysis. Each experiment was conducted three times to ensure accuracy and reliability.
2.6 Target selection and primer designPrimers were designed to target a specific 196 bp region within the XALB1 albicidin pathotoxin biosynthesis gene cluster between positions 43,128 and 43,323 of Xanthomonas albilineans (GenBank accession no. AJ586576.1) (National Center for Biotechnology Information, 1988). The selection was based on the crucial role that albicidin plays in the pathogenesis of Xalb, as previously highlighted (Royer et al., 2004; Pieretti et al., 2009). Additionally, this region was not homologous to any other species sequenced to date in the GenBank database.
For LAMP analysis, six primers were designed to recognize a total of eight distinct regions within the target sequence (Table 2 and Figure 1). These included two loop primers (XalbLF and XalbLP), two outer primers (XalbF3 and XalbB3), and two inner primers (XalbFIP and XalbBIP). The design process was carried out using the NEB LAMP primer designing tool available at https://lamp.neb.com. For qPCR analysis, forward and reverse primers (XalbFP and XalbRP) (Table 2) were designed using the NCBI primer blast web tool (Ye et al., 2012). To ensure the specificity of each primer, the corresponding sequences were screened against the NCBI nucleotide and genome databases using the BLASTn tool (Chen et al., 2015). All primer sequences showed 100% homology to the corresponding Xalb sequences. Furthermore, the possibility of hairpin and dimer formation was assessed using the OligoAnalyzer™ Tool provided by Integrated DNA Technologies Inc., United States to ensure the primer design robustness and avoid any potential issues with primer self-interactions.
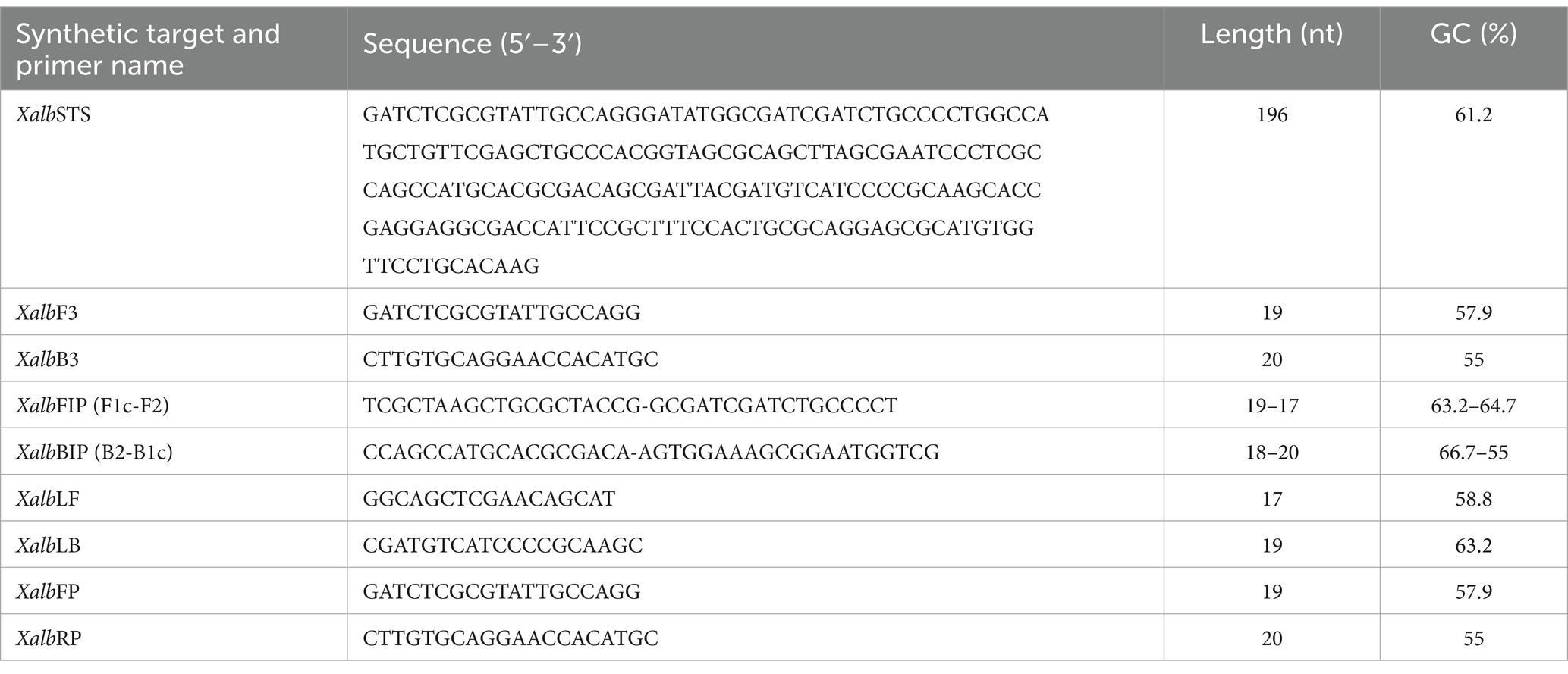
Table 2. Details of primer sets designed to develop Xalb-specific LAMP & qPCR assay.
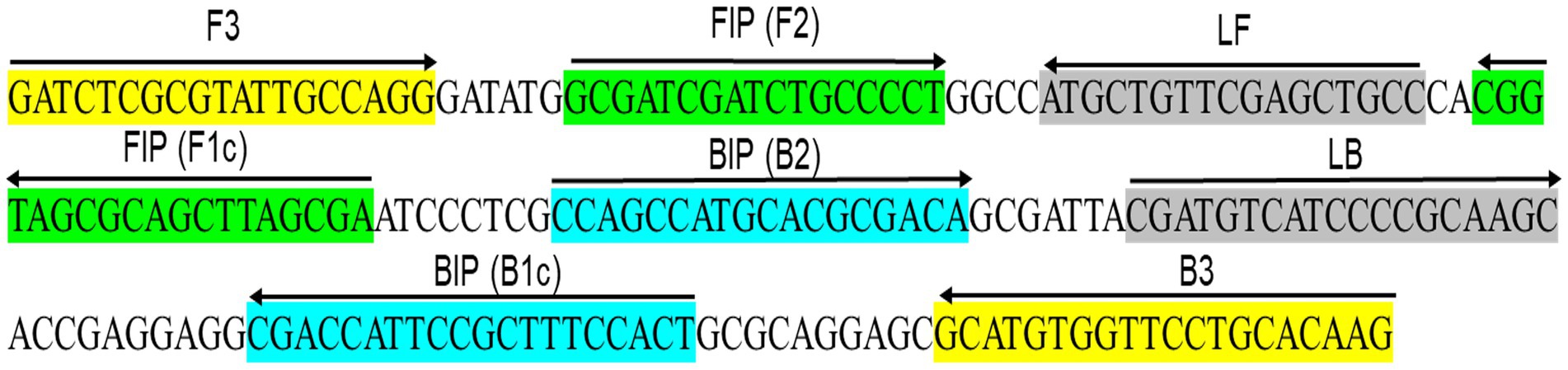
Figure 1. Schematic illustration depicts the primer design for the proposed LAMP assay, showing the locations of the eight primers that span the Xanthomonas albilineans gene cluster XALB1 sequence (GenBank Accession No. AJ586576.1). The right arrow indicates the sense sequence of the primer, while the left arrow represents its complementary sequence.
2.7 LAMP reaction conditionsThe standard LAMP mixture conditions were followed (Yang et al., 2022), with slight adjustments following the manufacturer’s instructions. For WarmStart fluorescent LAMP/RT-LAMP reactions, a 25 μL mixture was prepared, consisting of 2.5 μL of a 10X LAMP primer concentration (2 μM each of XalbF3 and XalbB3, 16 μM each of XalbFIP and XalbBIP, and 4 μM each of XalbLF and XalbLB). To this, 12.5 μL of 2X WarmStart Multi-purpose master mix, 0.5 μL of SYBR Green fluorescent dye, 2 μL of DNA template, and 7.5 μL of nuclease-free water were added. For the fluorescent LAMP assay, real-time amplification was performed at 65°C for 40 min using a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories Pty Ltd., Australia). In this LAMP assay, fluorescence threshold time (Tt) is used to quantify amplification, analogous to Cq (quantification cycle) in qPCR. Unlike qPCR, LAMP amplification is isothermal and proceeds continuously, yet Tt represents a measurable and reproducible point where fluorescence exceeds a defined threshold (Schmidt et al., 2021). For WarmStart colorimetric LAMP reactions, a 25-μL mixture was prepared, comprising 2.5 μL of a 10X LAMP primer concentration (2 μM each of XalbF3 and XalbB3, 16 μM each of XalbFIP and XalbBIP, and 4 μM each of XalbLF and XalbLB). To this, 12.5 μL of 2X WarmStart colorimetric LAMP master mix, 2 μL of DNA template, and 8 μL of nuclease-free water were added. The mixture was incubated at 65°C for 40 min using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories Pty Ltd., Australia). Subsequently, the amplified products were stored at 4°C prior to further analysis. The colorimetric LAMP products were visually inspected, with samples turning yellow classified as Xalb-positive, and those remaining pink considered Xalb-negative, following the manufacturer’s instructions (New England Biolabs, United States). Sterile distilled water (ddH2O) or fresh sap were used as no-target controls (NTCs). Meanwhile, Leifsonia xyli subsp. xyli (Lxx) cells or purified DNA from Lxx cells were utilized as negative controls (NG). Each sample, NTC or NG was analyzed in triplicate to evaluate intra-assay variability which was determined by calculating the SD (standard deviation) between the Tt (threshold time) values for each replicate.
2.8 Validation of qPCR assay and gel documentationThe qPCR experiments were conducted following the guidelines provided by the manufacturer (New England Biolabs, United States). Each qPCR mixture, with a volume of 20 μL, consisted of 10 μL of 2X SensiFAST SYBER No-ROX Mix, 0.8 μL each of XalbFP (10 μM) and XalbRP (10 μM), 2 μL of DNA template, and 6.4 μL of nuclease-free water. To perform the qPCR, a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories Pty Ltd., Australia) was utilized, applying the following reaction conditions: initial denaturation at 100°C for 1 min, 40 cycles at 98°C for 15 s, 52°C for 30 s, and 72°C for 30 s. The reaction was then terminated with a heating step at 72°C for 2 min, followed by a hold at 4°C for 5 min. At the completion of the reaction, the Cq value for each dilution was analyzed. A positive result for Xalb was recognized if it was observed within less than 40 cycles. Each assay was performed in triplicate for each repetition to ensure accuracy and reliability. For gel documentation, 5 μL of each LAMP or PCR product was loaded onto a 1% agarose gel and electrophoresed in 1X Tris-acetate-EDTA (TAE) buffer at 90 V for 40 min. The gel was then stained with SYBR Safe and visualized using UV light on a gel documentation system. The sizes of the amplified products were estimated using a 100 bp GeneRuler (Thermo Fisher Scientific, Australia).
2.9 Statistical analysisData analyses were conducted using OriginPro 2022 v.9.9.0.225 (OriginLab, Northampton, Massachusetts, United States), the R programming language (version 4.2.1), and Microsoft Excel 365 (United States). To analyze the sensitivity, serial dilutions of known Xalb target sequence, Xalb cells, and purified genomic DNA concentrations were utilized to create a standard curve for the absolute quantification of Xalb. This standard curve was represented as a semi-log regression line plot, with Tt and Cq values plotted against the (−log) of the input DNA template amount. The efficiency (E) of the fluorescence LAMP and qPCR assays was calculated using the formula E = (10−1/slope) − 1. Both the fluorescence LAMP and qPCR results obtained with the target-specific primers were validated within the range of 0.9 < E < 1.1, with an E value closer to 1.0 indicating higher amplification efficiency. To estimate the development of bacterial population over time in leaf tissue, meristematic tissue, and xylem sap of two sugarcane cultivars (Q44 and Q208) using LAMP, a logistic model was used as:
where, P is estimated development of Xalb population, d is estimated maximum population, b is estimated rate of development, c is inflection point or estimated median Xalb population development time, and t is Xalb population development time (month).
To convert the LAMP data (Tt values) to Xalb population or Cells the following equation was utilized:
where, x is the logarithm of the initial cell number a is the y-intercept, b is slope of the standard curve obtained from the Xalb cell number, and y is the Tt value.
To predict the cultivar disease ratings for Xalb, Tt and Cq values from the analysis of sugarcane samples were considered, whereby a correlation between Cq values and pathogen titer, was previously shown to correspond to disease ratings of cultivars (Davis et al., 1988; Rott et al., 1997). To analyze the Tt and Cq datasets, a linear mixed model was fitted using proc mixed in SAS version 9.4 (SAS Institute, Cary, NC). This model enabled the evaluation and interpretation of Tt and Cq data in relation to the disease ratings of sugarcane cultivars. Cultivars were considered as fixed effects, while block (replication) and the error term (residual) were regarded as random effects in the statistical analysis. To determine appropriate significance factors, protected-mean comparisons of all possible pairwise differences in Tt and Cq values were analyzed at alpha = 0.05 using Fisher’s protected LSD test. The PDMIX800 SAS Macro was employed to convert the mean separation outputs into letter groupings (Saxton, 1998). Spearman rank correlation was conducted to assess the relationship and accuracy of the newly developed method by comparing the outputs of the LAMP and qPCR techniques. Data visualizations were created using BioRender (BioRender, 2022), SnapGene software, and Microsoft PowerPoint 365 (United States). The color change observed in the colorimetric LAMP reaction was captured using a mobile camera (Samsung Galaxy A52s).
3 Results 3.1 Validation of novel Xalb target sequences and assessment of LAMP assay robustnessThe newly designed LAMP primer sets successfully amplified the targeted XALB1 albicidin pathotoxin biosynthesis gene cluster region (196 bp) in both colorimetric and fluorescent LAMP assays using 10 pM (equivalent to 107 copies/μL) of synthetic target sequences. This confirmed the efficacy of the primers in detecting Xalb (Supplementary Figure S1A). In the colorimetric LAMP assay, a distinct color change from pink to yellow indicated a positive response when Xalb targets were present, while the no-template control (NTC) and negative control (NC) remained pink (Supplementary Figure S1B), highlighting the specificity of the primers and the suitability of the assay for naked-eye detection of Xalb. In the gel electrophoresis, the LAMP amplification products exhibited a typical ladder-like pattern, with the primary band corresponding to the expected 196 bp amplicon, along with other bands resulting from the amplification properties of LAMP (Supplementary Figure S1C).
Furthermore, the positive color change to yellow was consistently observed across all eight serially diluted concentrations of synthetic target sequences, from 107 to 100 copies/μL, with the lowest detection limit of this assay of 1 copy/μL (1 ag/μL) (Figure 2A). Similarly, in the fluorescent LAMP assay, detection of the synthetic targets was achieved solely in the infected sap samples, with no signal observed in the NTC (Supplementary Figure S2). The fluorescent LAMP signal exhibited a direct and reproducible correlation with the target DNA concentration, as indicated by the Tt values obtained at various time points throughout the reaction (Figure 2B), yielding a high correlation coefficient (r) of 0.99. The lowest detection limit for the LAMP assay remained at 1 copy/μL (1 ag/μL), and gel electrophoresis confirmed the amplification of the expected 196 bp product, along with additional bands, exclusively in positive reactions (Figures 2A; Supplementary Figure S2).
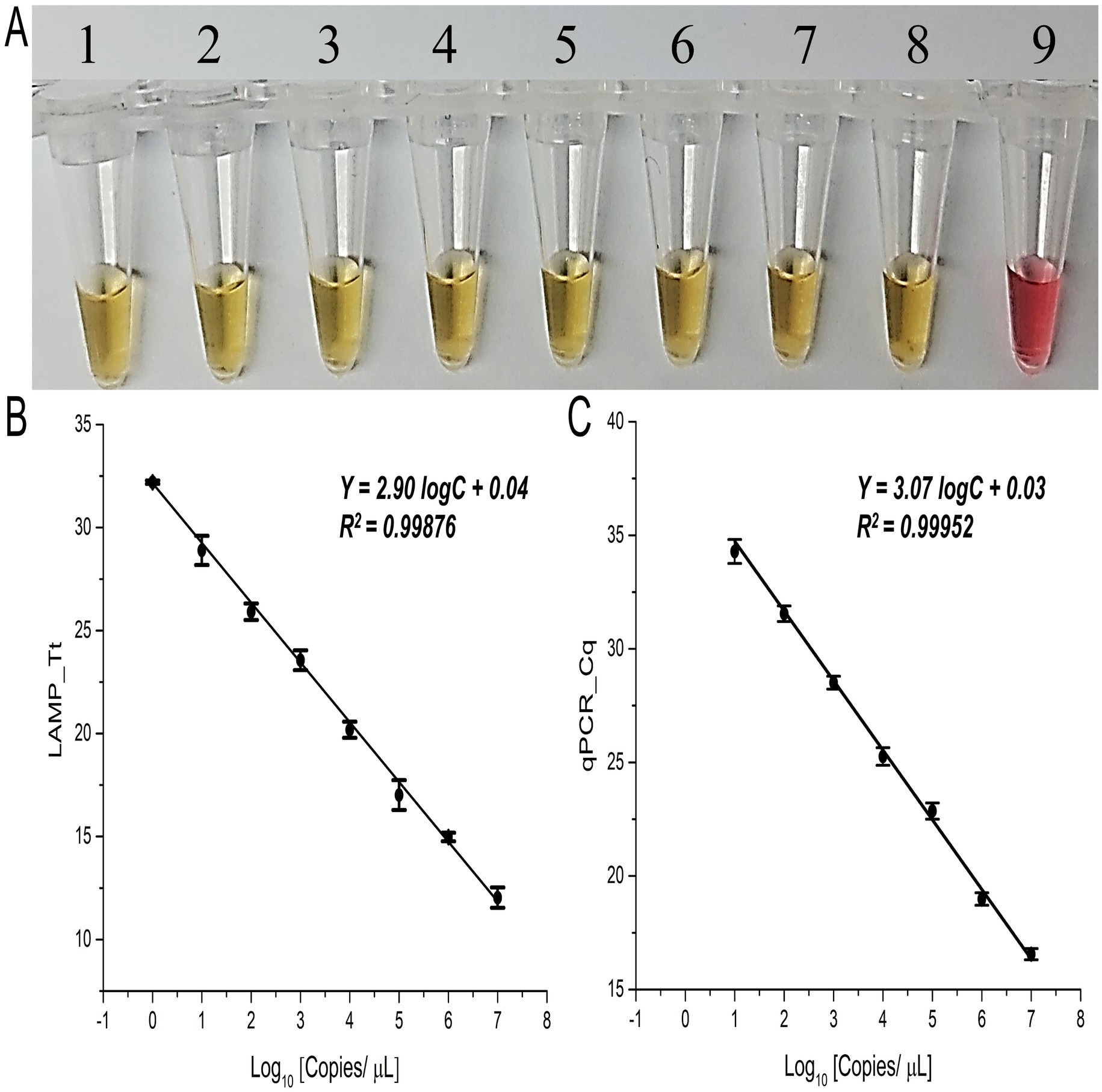
Figure 2. Validation of LAMP and qPCR primers with synthetic target concentrations. (A) Colorimetric LAMP detection for the specified concentrations; tubes 1 to 8 show 1:10 dilutions of the synthetic target (107–100 copies/μL, or 10 pg/μL–1 ag/μL); tube 9 is the no target control (NTC). (B) Fluorescent LAMP Tt values and (C) qPCR Cq values for the specified synthetic target concentrations (107–100 copies/μL, or 10 pg/μL–1 ag/μL). Error bars indicate the standard deviation from three biological replicates.
To further validate the results of the LAMP assay, a standard curve for qPCR was established using titrated synthetic targets ranging from 107 to 100 copies/μL of Xalb (Figure 2C), with a lowest concentration detected was 10 copies/μL (100 ag/μL) or 1 nM at an annealing temperature of 52°C (Supplementary Figure S5A), which was 10 times less sensitive than the LAMP assay (Figure 2B). Moreover, a single amplicon of the correct size (196 bp) was visualized exclusively from the positive samples (Supplementary Figures S5B, S6B), further supporting the robustness and reliability of the LAMP assay for detecting Xalb.
3.2 Validation of LAMP-based in-situ detection and quantification method for XalbUsing the standard curve generated from predetermined titrated concentrations of Xalb cells (107–100 cells/μL), the lowest detection limit of the colorimetric LAMP assay was approximately 1 cell/μL (Figure 3A), consistent with the fluorescence LAMP assay results (SD ≤5% across three replicates, r = 0.99; Figure 3B). Additionally, the expected LAMP amplicon of 196 bp, along with a ladder-like pattern, was visible on agarose gel for positive samples only (Supplementary Figure S3).
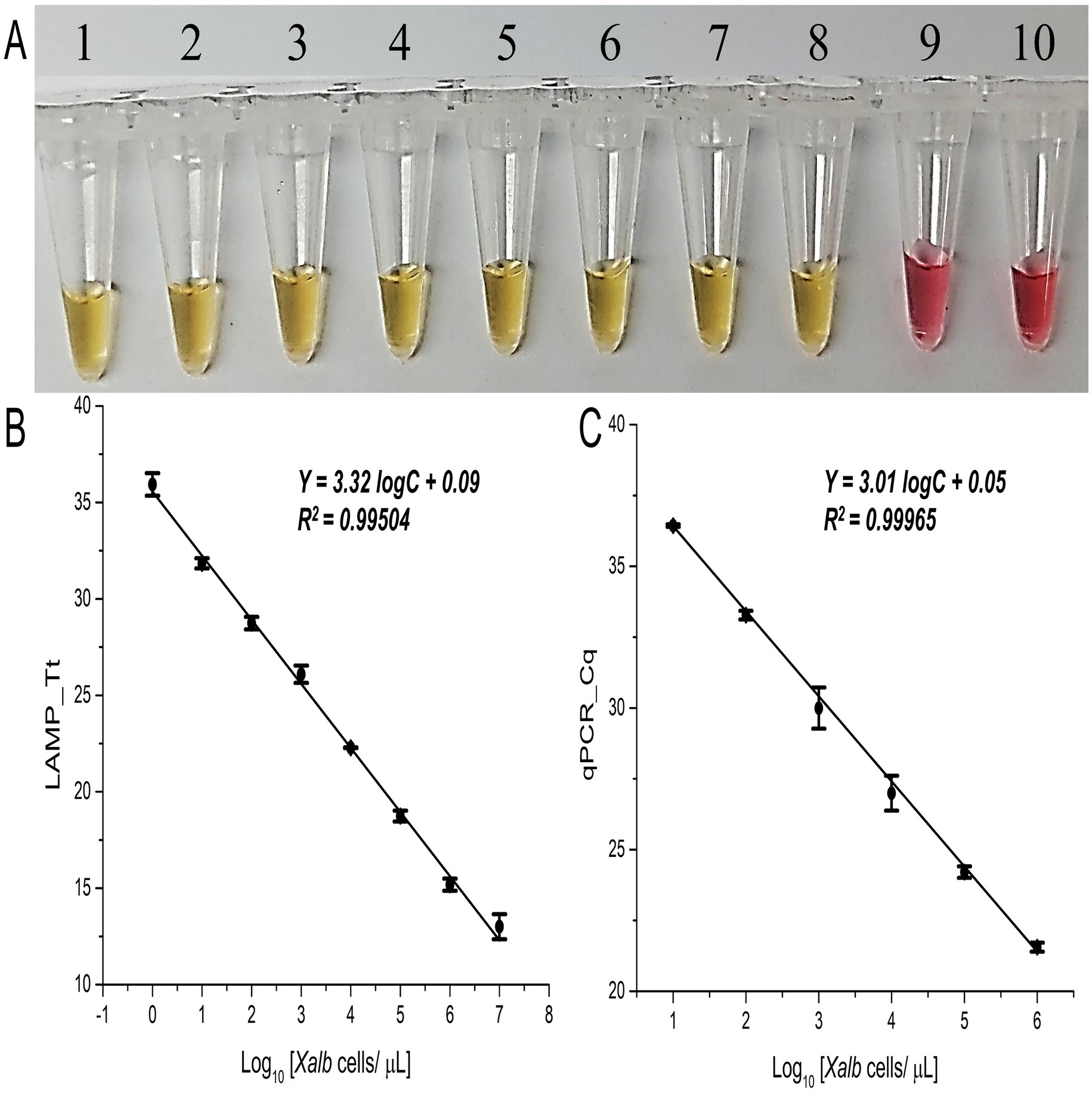
Figure 3. Sensitivity and specificity analysis of the LAMP and qPCR assays. (A) Colorimetric LAMP detection for the specified samples; Tubes 1 to 8 contain 1:10 dilutions with Xalb cells spiked into fresh, clean sap (107–100 cells/μL); Tube 9 is the No Target Control (NTC); Tube 10 contains a known concentration of spiked Lxx cells (107 cells/µL). (B) Tt values from fluorescence LAMP, and (C) Cq values from qPCR detection for the specified concentrations of Xalb cells spiked into fresh xylem sap (107–100 cells/µL). Error bars indicate the standard deviation across three biological replicates.
To further validate the LAMP assay with Xalb cell samples, a qPCR standard curve was created by plotting Cq values against the log-transformed quantities of purified DNA extracted from titrated Xalb cell concentrations. The qPCR assay reliably amplified the target region from as few as 100 Xalb cells (Figures 3C; Supplementary Figure S6B), with the expected 196 bp amplicon observed on agarose gel for positive samples (Supplementary Figure S6D). A strong correlation (r = 0.99) was observed between Tt and Cq values from LAMP and qPCR assays. Notably, the LAMP assay showed a sensitivity 100 times greater than qPCR for in-situ detection and quantification of Xalb bacteria (Figure 3B vs. Figure 3C).
3.3 Detection of Xalb in different sugarcane samplesTarget DNA was amplified from all the field samples collected from the first LS field trial, indicating varying levels of pathogen load (Figures 4, 5). In colorimetric LAMP, a yellow color development was observed in all field samples, providing a rapid and consistent qualitative indication of bacteria in the sample (Figure 4).
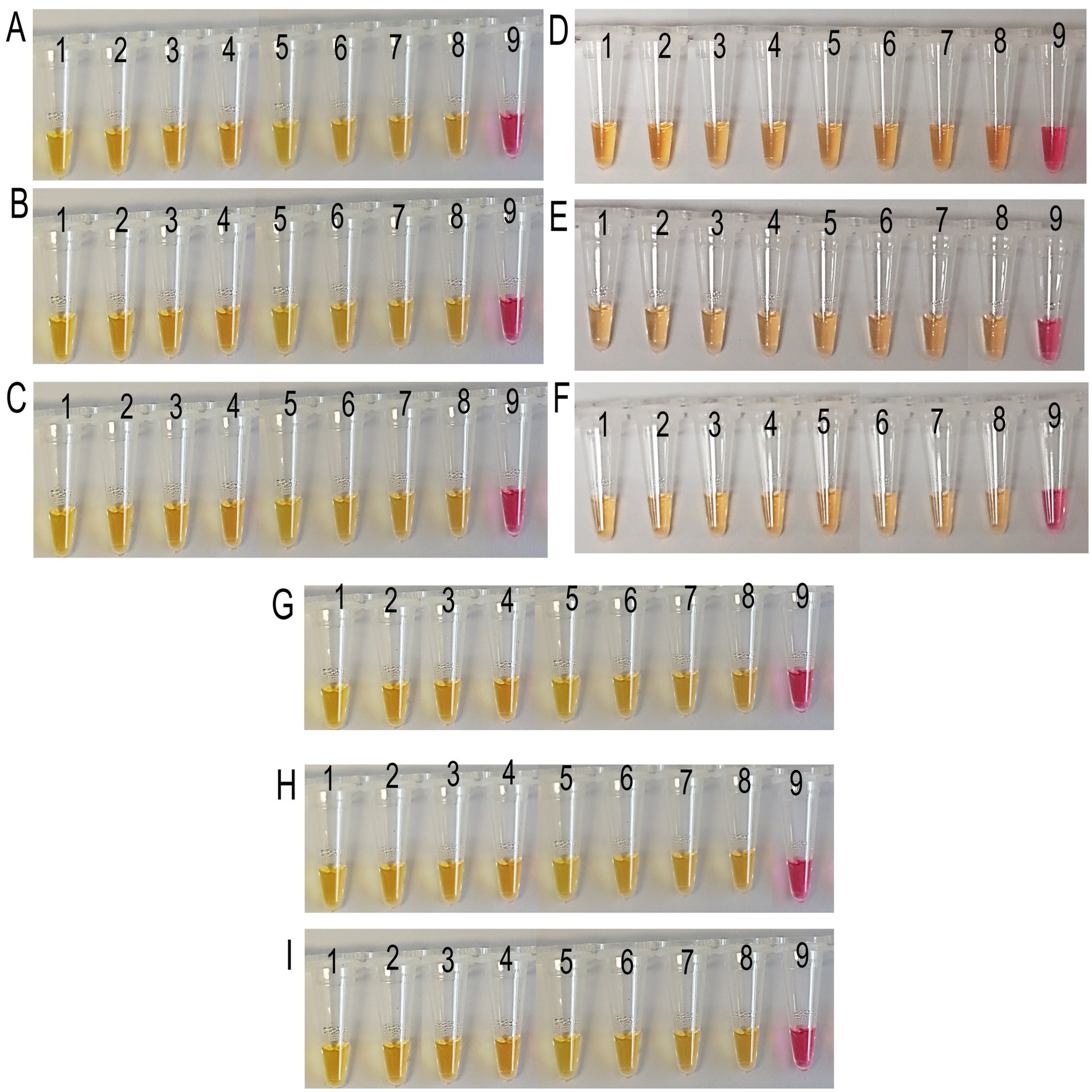
Figure 4. Field application of LAMP colorimetric assay for all the analyzed sugarcane samples collected from SRA Woodford LS screening trials. (A–C) Xylem sap, leaf tissue, and meristematic tissue samples collected in July 2022. (D–F) Xylem sap, leaf tissue, and meristematic tissue samples collected in October 2022. (G–I) Xylem sap, leaf tissue, and meristematic tissue samples collected in January 2023. Where, tubes 1 to 8: LS-infected xylem sap samples-Q87, Q63, Q68, Q208, Q96, Q124, Q44, and Q133; tube 9: no target control (NTC).
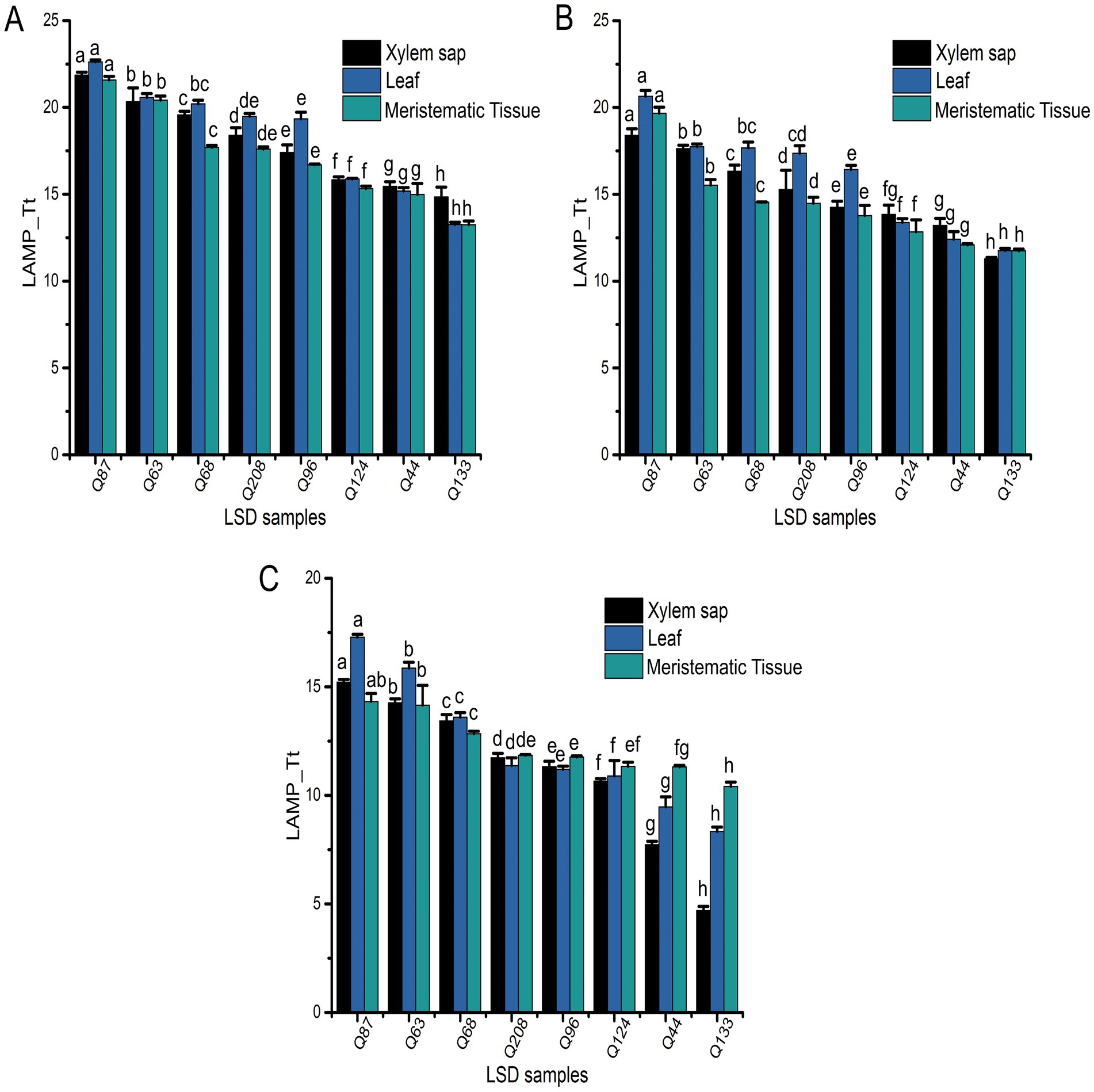
Figure 5. Field application of LAMP Fluorescence assay for all the analyzed field collected samples from the SRA Woodford LS trial sites. Obtained LAMP-Tt values of xylem sap, leaf tissue, and meristematic tissue samples collected in (A) July 2022, (B) October 2022, or (C) January 2023. Each bar is the mean of three replications, and bar associated with the same letter for a particular sample type is not significantly different according to Fisher’s protected LSD test (p = 0.05).
The fluorescent LAMP assay effectively amplified Xalb DNA from all collected samples across different Tt values, reflecting varying levels of pathogen load (Figures 5A–C). Most samples exhibited Xalb DNA concentrations within the assay’s lowest detection limit of 1 aM, highlighting its suitability for practical field applications. The detected Xalb DNA level in the different sugarcane samples correlated with bacterial load. For example, the Q87, Q63, Q68, and Q208 cultivars showed relatively higher Tt values (11.36–22.62) for all the analyzed sample types collected at different time points, suggesting lower Xalb presence (approximately 104–107 cells/μL) in xylem sap, leaf tissue, and meristematic tissue (Figures 3B, 5A–C). Conversely, cultivars Q44, Q96, Q124, and Q133 displayed progressively lower Tt values (4.69–19.33), indicating higher bacterial load (around 107–109 cells/μL) in these samples (Figures 3B, 5A–C). This data demonstrates that the LAMP method is effective for detecting and quantifying Xalb titres in infected sugarcane samples across various cultivars, irrespective of their resistance status. Notably, significantly different amounts of the amplified product of expected size (196 bp) for Xalb DNA were detected among them, with no amplification observed in control reactions (Supplementary Figure S4).
The target qPCR region was successfully amplified from all infected field samples (Figure 6), revealing a significant correlation (r = 0.9, p < 0.001) between the qPCR values and the results of the LAMP assay, as outlined in Table 3. Despite the differences in amplification properties between LAMP and qPCR, strong correlations (r > 0.9) were observed between LAMP and qPCR results (Table 3), regardless of sample collection date or site. Additionally, a single amplicon was observed on the gel from positive samples only (Supplementary Figure S7), confirming the specificity of the assay.

Figure 6. Field application of qPCR assay for all the analyzed sugarcane samples collected from the SRA Woodford LS screening trials. Obtained qPCR-Cq values of xylem sap, leaf tissue, and meristematic tissue samples collected in (A) July 2022, (B) October 2022, or (C) January 2023. Each bar is the mean of three replications, and bar associated with same letter for a particular sample type is not significantly different according to Fisher’s protected LSD test (p = 0.05).
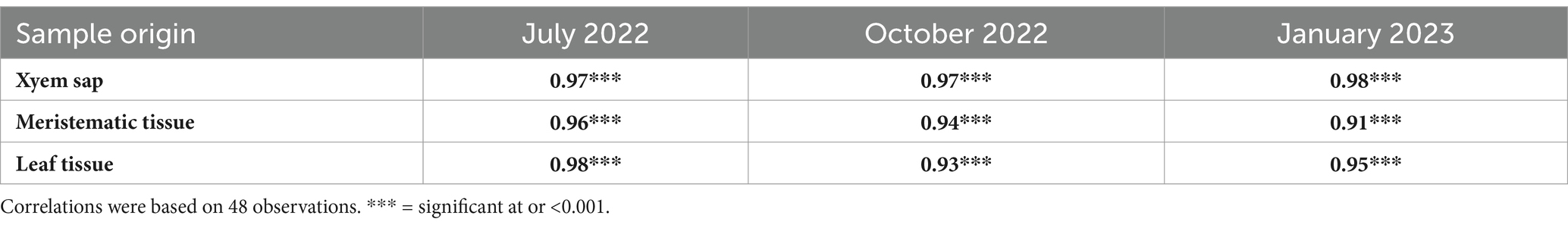
Table 3. Spearman correlation coefficients determined between LAMP and qPCR data collected from different parts of sugarcane plants and at three different post-inoculation collection time points.
When analyzing the Tt values from the LAMP experiment across different sample types using the SAS Proc Mixed Model, significant differences (p < 0.0001) were observed in Tt values (indicating Xalb load) among clones, sample origins (leaf tissue, meristematic tissue, or xylem sap), sampling times, or their interactions (Supplementary Table S1). Strong correlations were identified between leaf tissue samples and disease ratings when collected at 5 months (r = 0.75) and 8 months (r = 0.8) post-inoculation. However, no differences in Tt values were found between cultivars when samples were taken 11 months after inoculation (Figure 7A). A moderate correlation (r = 0.53) was observed between varietal resistance and Tt values at around 8 months post-inoculation, while low or no correlation was found at 5- or 11 months post-inoculation (Figure 7B). Additionally, there was no meaningful relationship between varietal resistance and Tt values in xylem sap (Figure 7C). These results suggest that leaves may be the suitable plant part for effective sampling for leaf scald diagnosis and varietal disease rating.
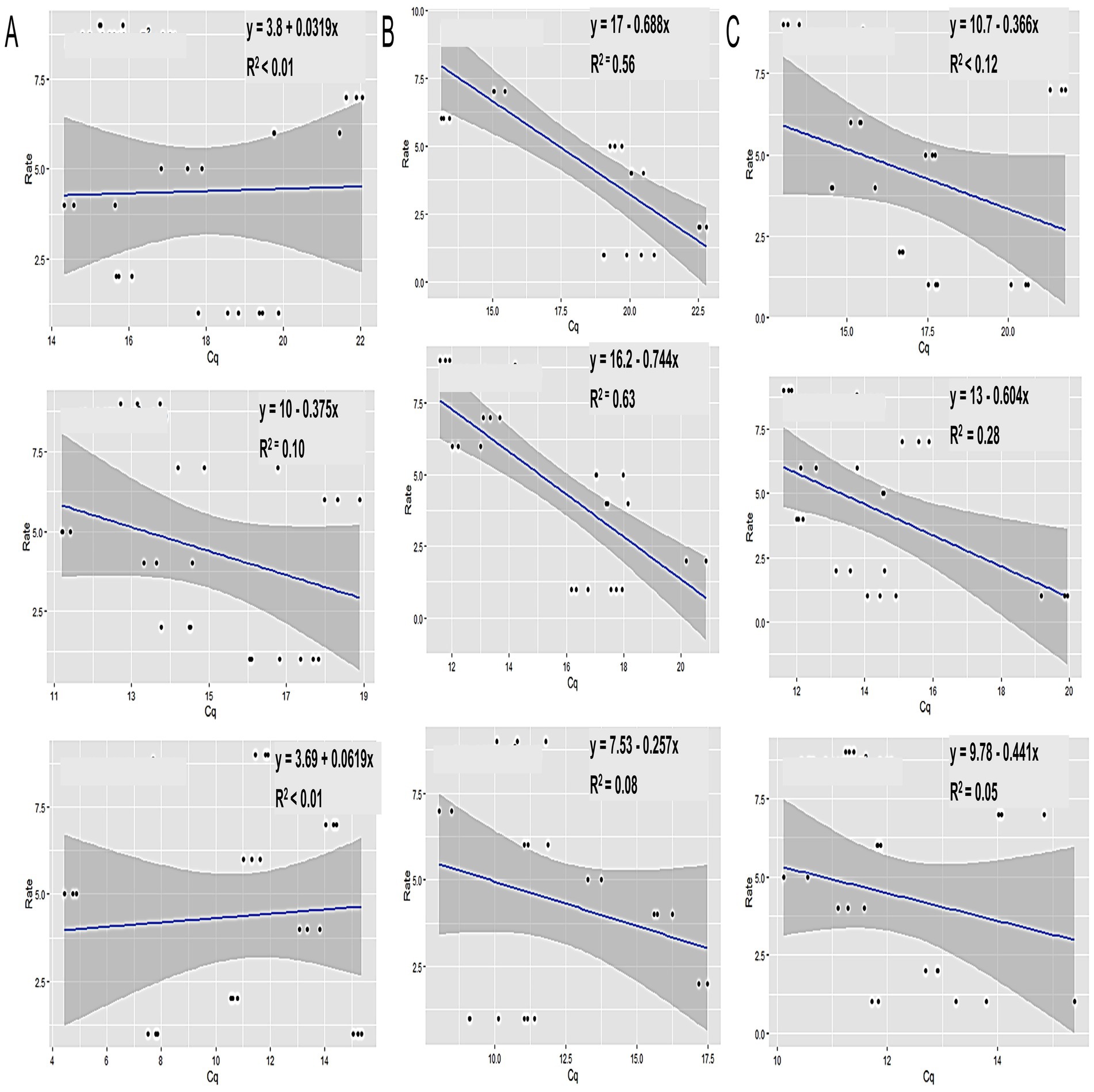
Figure 7. The relationship between LS disease ratings (rate) and Tt values. (A) Xylem sap, (B) leaf tissue, and (C) meristematic tissue samples collected from sugarcane cultivars with various scales of disease rating (1 to 9), inoculated by decapitating and sprayed with Xalb after planting. Samples were taken at 5 (top), 8 (middle), and 11 (bottom) months after inoculation, respectively.
3.4 Identification of the most suitable leaf/leaves tissue for samplingSignificant differences were observed in Tt and Cq values among cultivars collected from the second LS trial and between the methods used (LAMP and qPCR). Specifically, in the interaction between cultivar and method (p < 0.001) and between the two methods themselves (p < 0.001) (Table 4). However, no significant differences were found between leaf locations within a variety, indicating a similar Xalb load among the leaf positions (TVD, TVD +1 and TVD −1) on a sugarcane stalk (Table 4).
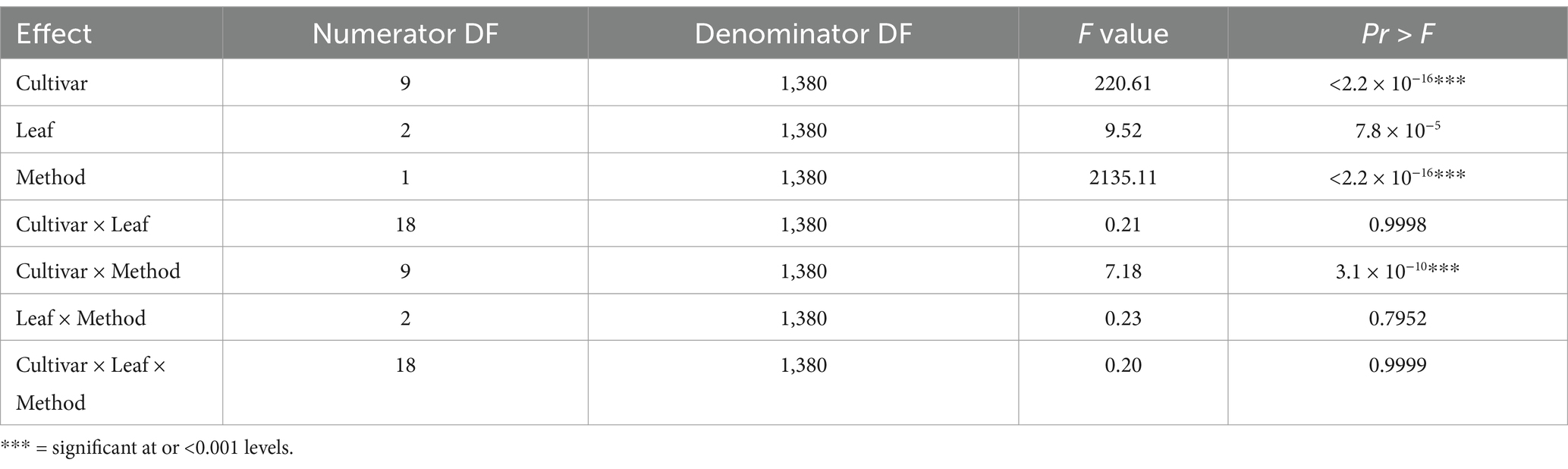
Table 4. Type 3 test of fixed effects of cultivar and interactions.
3.5 Development of Xalb populations in various sugarcane samples over timeFollowing inoculation, the development of the Xalb population over time in leaf tissue, xylem, and meristematic tissues were analyzed for two sugarcane cultivars collected from the third LS trial, described by the regression equation in Table 5 and illustrated in Figure 8. The initial Xalb population, as estimated from the regression equations, was approximately 2.4 × 103 Cells/μL for the susceptible cultivar Q44 and less than 60 Cells/μL for the resistant cultivar Q208. The maximum Xalb levels in the leaf tissue of Q208 and Q44 were estimated to be approximately 1.1 × 108 Cells/μL and 1.8 × 109 Cells/μL, respectively, indicating that the levels in Q44 were more than 17 times higher than those in Q208. Indeed, the rate of Xalb reproduction was slower in Q208 compared to Q44; taking about 146 days for Q208 to reach 50% of its maximum Cells, while Q44 reached it in approximately 131 days. Similar trends were observed among the meristematic tissues and xylem saps between the two cultivars.
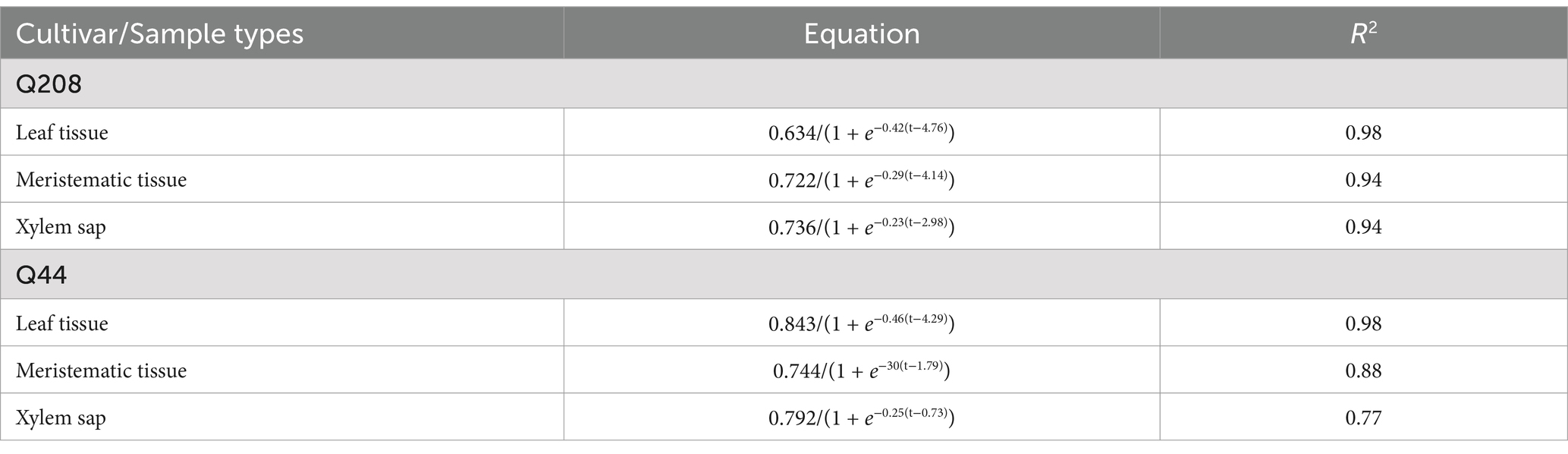
Table 5. Regression equation describing Xalb population development over time in leaf tissue, meristematic tissue, and xylem sap samples.
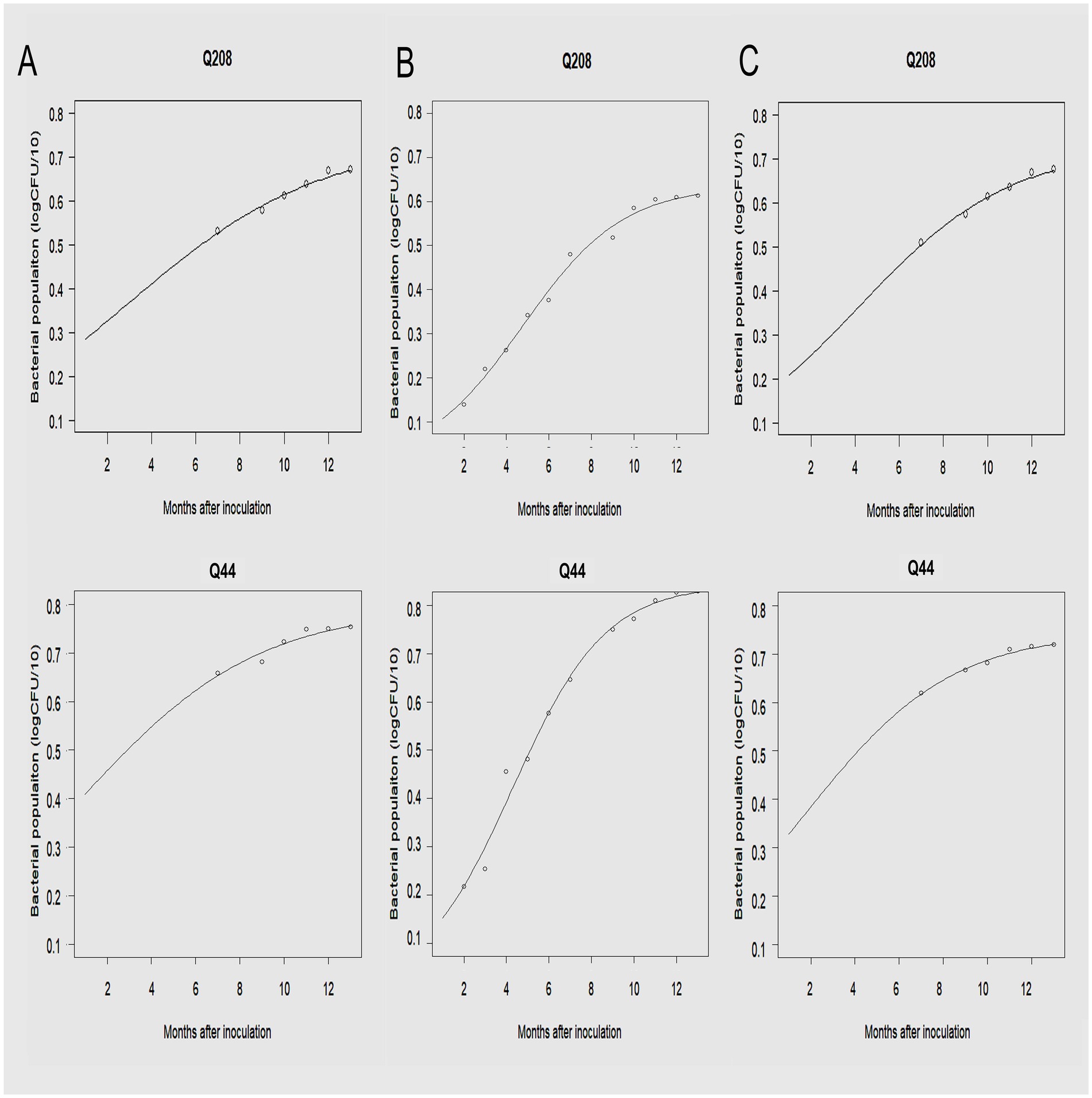
Figure 8. The development of Xalb population on a resistant (Q208) and a susceptible (Q44) cultivar in (A) xylem sap, (B) leaf tissue, and (C) meristematic tissue of sugarcane plants over time.
4 DiscussionThis proof-of-concept study developed a highly specific and sensitive LAMP detection method for the quantification of Xanthomonas albilineans (Xalb) in sugarcane. The amplification mechanism of LAMP differs from qPCR by operating isothermally without discrete cycles. Instead, LAMP uses threshold time (Tt), the point at which fluorescence exceeds a set threshold, to quantify template concentration (Schmidt et al., 2021). Our study shows that Tt reliably correlates with initial template concentration under optimized conditions (Njiru et al., 2008), similar to the cycle quantification threshold (Cq) of qPCR. Previous LAMP and PCR protocols described in the literature targeted the ITS region, the albI gene, and the XAF DNA fragment. However, these methods often amplified anonymous fragments from Xalb genomic DNA (Dias et al., 2018; Garces et al., 2014; Gutierrez et al., 2016; Jensen et al., 1993; Wang et al., 1999; Welsh and McClelland, 1991). Existing primers (L1/G1, T3A/T5A, XAF1/XAR1, XAprF6/XAprR6, and FIP/BIP, F3/B3) were found to lack specificity for Xalb, as they also detected saprophytic bacteria from sugarcane (Pieretti et al., 2015). To address this limitation, we designed primers (Table 2) targeting the XALB1 albicidin pathotoxin biosynthesis gene cluster, optimizing specificity and efficiency. Primer efficiency was assessed based on the intensity of amplification products of the expected and specific 196-bp target bands (Supplementary Figures S3, S4). The specificity of three primer pairs was further validated by testing their ability to detect Xalb in the Saccharum genus and other sugarcane pathogens such as Lxx (Figure 3; Supplementary Figure S3). Results confirmed the strong specificity of the designed primers, as they consistently produced specific amplification products (196 bp). However, additional testing with related pathogens and sugarcane isolates is necessary to ensure the LAMP assay’s efficacy across other Xalb serogroups or haplotypes.
A near-ideal amplification efficiency (slope ≈ 3) observed in the florescence LAMP standard curve (Figures 2B, 3B) which reflects the high reproducibility of the assay under optimized conditions. Although LAMP operates differently from qPCR, continuous amplification can produce a log-linear relationship when fluorescence thresholds are consistently defined. The efficiency highlights the optimized conditions, such as precise temperature control, primer design, and consistent fluorescence thresholding, supporting LAMP’s potential for quantitative applications (Mori et al., 2004). Our LAMP assay demonstrated higher sensitivity than conventional qPCR, detecting Xalb at 100-fold lower cell concentrations compared to qPCR. This sensitivity surpasses previous reports of Xalb detection by LAMP using purified DNA (10 Cells/mL) (Dias et al., 2018). While qPCR and nested-PCR methods have been shown to achieve low detection limits (102–103 Cells) for Xanthomonas albilineans, they are limited in their ability to determine pathogen population densities directly (Garces et al., 2014; Dias et al., 2018; Wang et al., 2020). Additionally, qPCR requires sophisticated and costly equipment, as well as labor-intensive sample preparation to mitigate reaction inhibition from contaminants. Nested PCR involves an extra gel electrophoresis step, adding to the complexity and time required. In contrast, this study highlights the compatibility of a reagent-free, heat-induced DNA extraction method with LAMP, enabling a simple, rapid, and visually detectable diagnostic solution with quasi-quantitative capabilities for Xalb. This builds on prior findings that bacterial cells are vulnerable to high-temperature damage (Jose and Brahmadathan, 2006; Umer et al., 2021). By eliminating the need for multistep sample processing and expensive commercial kits, our approach offers a cost-effective and accessible alternative, suitable for practical applications in field settings.
One of the challenging aspects of leaf scald disease (LS) is that many sugarcane plants infected with Xalb do not exhibit symptoms (Rott et al., 1997; Gutierrez et al., 2016), or the symptoms are so subtle that they escape detection (Rott et al., 1988; Ricaud and Ryan, 1989). In this study, we evaluated the detection efficiency of our assay by analyzing various sugarcane samples (leaf tissue, meristematic tissue, and xylem sap) collected from 10 cultivars 2–3 months post-inoculation. The successful detection of Xalb in field-derived samples from asymptomatic stalks confirmed the high efficiency of our assay for identifying Xalb in contaminated field samples, as previously demonstrated for LAMP (Dias et al., 2018). Our findings showed that bacterial populations in leaf tissue are a more reliable indicator of the genetic resistance of sugarcane varieties to LS compared to other sample types, which aligns with traditional assessments of LS resistance based on disease incidence and severity. A strong correlation between Xalb populations in the shoot apex and disease severity, as reported by Rott et al. (1997), underscores the value of accurate bacterial quantification for resistance screening. Traditionally, resistance screening has relied on the erratic expression of symptoms following inoculation (Rott and Davis, 2000). This study demonstrated that LA
留言 (0)