
記住我
Intellectual disability (ID) includes a variety of developmental disorders defined by limitations in cognitive abilities and adaptive behavior according to the diagnostic and statistical manual of mental disorders, 5th Edition (1). An intelligence quotient (IQ) under 70 implies a deficit in intellectual functioning, which in combination with adaptive functioning determines further classification as severe, or profound, moderate and mild. ID can manifest in varying degrees of severity and is often accompanied by challenges in learning, communication, and daily life skills. The onset of ID typically occurs before the age of 18 and affects approximately 1–3% of the global population (2–4). ID is primarily limited to individuals over 5 years old, while global developmental delay (DD) is used for children 5 years old or younger (5, 6).
ID may arise from both genetic and non-genetic factors. Non-genetic causes include nutritional deficiencies, exposure to toxic substances, maternal infections during pregnancy, hypoxic–ischemic events, brain radiation, encephalitis and traumatic brain injuries (7). However, a significant proportion of ID cases are attributed to genetic causes, such as chromosomal abnormalities including trisomies, deletions, and duplications (8, 9). Genetic syndromes associated with ID frequently present with additional clinical symptoms, including motor, psychiatric, and sensory impairments (10). These co-occurring conditions can make diagnosis more difficult and may hinder the correct identification. The children with congenital hearing loss may face developmental delays that resemble conditions like autism (11). Furthermore, sensory impairments like hearing loss may be mistaken for behavioral issues (12), complicating both diagnosis and treatment. Advances in genomic technologies, including array-comparative genomic hybridization (Array-CGH) and WES, have enabled the identification of many genetic variants linked to ID (13, 14). The discovery of these genes reveals its underlying causes and expands our understanding of the disease.
Among these genes, ASH1L (Absent, small, or homeotic1-like) has garnered significant attention due to its critical role in brain development. ASH1L (also referred to as KMT2H, ASH1-like, ASH1L1, ASH1, or huASH1) is a enzyme classified as a histone-lysine N-methyltransferase and encoded by the ASH1L gene which is located at chromosomal band 1q22 (15). As a member of the trithorax group family, ASH1L facilitates the methylation of specific histone lysine residues and lays a key role in regulating transcription and chromatin remodeling (16, 17). Mutations or loss of function in ASH1L were linked to various developmental disorders, including ID and ASD. These mutations typically result in a nonfunctional enzyme, disrupting histone methylation and altering gene expression, which in turn affects brain development. First described ASH1L variant in patients in clinical study involving three patients with ID or ASD due to de novo ASH1L missense variants (18–20). In addition, ASH1L mutation have also been linked to seizures, which broadens the diversity of both genetic and clinical features seen in ASH1L-related neurodevelopmental disorders (21). ASH1L mutations result in altered methyltransferase enzyme activity and changes in neuronal morphology, leading to cognitive impairments and disruption of ASH1L’s regulatory functions (22). Mutations within the ASH1L PHD-BAH domain may disrupt interaction with methyltransferase enzyme in vitro, potentially compromising chromatin remodeling (23). Mouse models with ASH1L exon 4 deletion resulting in a premature stop codon (p.V1693Afs*2) exhibit abnormal cortical neuron differentiation and craniofacial abnormalities, providing insights into the molecular mechanisms underlying ASH1L-associated neurodevelopmental disorders (24). While previous reports have identified several heterozygous loss-of-function (LOF) variants in ASH1L, such as nonsense, frameshift, and deletions, the phenotypic spectrum remains incompletely understood. In particular, the relationship between these mutations and the severity or specific features of the associated disorders is still being explored.
To address this gap, our study specifically aims to clarify the genotype–phenotype relationship of ASH1L variants, refine diagnostic criteria for ASH1L-associated neurodevelopmental disorders, and inform future research into targeted therapeutic strategies. Here, we identified a novel heterozygous nonsense variant of ASH1L, NM_018489.2: c.2479A > T (p.Lys827*), in a patient diagnosed with mild ID. This variant adds to the growing body of evidence supporting ASH1L’s role in neurodevelopmental disorders and expands the known phenotypic spectrum of this condition. Our findings emphasize the importance of larger-scale studies to further characterize the clinical impact of ASH1L nonsense variants and highlight the need for more comprehensive neurological evaluations.
Materials and methods SubjectsThe research informed consent was undersigned by the patient and parents. This study was carried out in accordance the ethics requirement. The study involved the proband and both parents. Family members were selected based on their relationship to the proband and symptoms of intellectual disability. The clinical assessment of Wechsler Adult Intelligence Scale (WAIS) was conducted prior to genetic testing to ensure thorough phenotypic characterization. The proband in our research is a 23-year-old female with a novel variant (c.2479A > T, p.Lys827*; NM_018489.3). She is the only child born to a non-consanguineous couple, and her prenatal, labor, and postnatal medical history are entirely normal, with no noted abnormalities. The proband has a score of 65 on the WAIS assessment, indicating borderline cognitive functioning. Additionally, it is reported that the proband has mild ID with poor learning abilities and poor memory. She can manage her daily life independently but shows slightly delayed responses. Her mother exhibits similar symptoms. Family history is important for the characteristics of ID on the mother’s side. The proband has no characteristics reported in other probands, for example, seizures. The proband is found to be non-dysmorphic. The results of Multiplex Ligation-dependent Probe Amplification (MLPA) test for the proband did not reveal any large segment variants within the detection range of the P070 kit. It is noteworthy that following clinical testing, this proband participated in a research study that uncovered four variants of unknown significance (VUS). The first is a missense variant in ACTL6B, which, according to OMIM, is associated with developmental and epileptic encephalopathies (OMIM#618468), and intellectual developmental disability with severe speech and walking deficits (OMIM# 618470). Another variant is a missense mutation in COQ8A, which has been linked to primary coenzyme Q10 deficiency (OMIM# 612016). The third is also a missense variant in DPP6 related to the intellectual developmental disabilities (OMIM# 616311). The last is an in-frame variant in SETD1B, which is associated with intellectual developmental disabilities accompanied by seizures and speech delays (OMIM# 619000).
Genomic DNA preparationWhole blood was collected in EDTA anticoagulant tubes (4 mL from the proband, 2 mL from parents and family members). Samples were processed and stored at −80°C to preserve DNA integrity during transportation to Kangxu Diagnostics (Beijing, China). Genomic DNA was extracted using the Qiagen FlexiGene DNA Kit, with quality assessed by Qubit 2.0 and NanoDrop 2000. We ensured ≥1.5 μg of DNA with a concentration of 50–100 ng/μL and OD 260/280 ratio of 1.8–2.0. DNA was fragmented into 180–280 bp segments, followed by end repair, A-tailing, adapter ligation, and exon capture using Agilent SureSelect Human ALL Exon V6 probes. Sequencing was performed on the T7 platform with 100× depth and PE150 reads. Libraries were validated using Qubit 2.0 and Agilent 2,100, with a minimum library concentration of 3 nM before sequencing.
Genetic analysis and ACMG integrationThe Ethics Committee of the Ganzhou Maternal and Child Health Hospital approved this research. Single-person WES was performed by Kangxu Diagnostics (Beijing, China). Given the proband’s complex phenotype, WES provided a more comprehensive genetic assessment, enhancing the likelihood of discovering relevant variants beyond well-characterized gene sets.Variants screening were based on clinical phenotypes of the affected subjects. Variants were filtered using biological information prediction tools (Polyphen2 Mutation Taster, SIFT and Splice Al), population database (ExAC, 1,000 Genome, dbSNP) and disease database (Clinvar., HGMD, OMIM). Thresholds for pathogenicity were determined based on ACMG guidelines, ensuring a standardized and reliable classification of variants. The ACMG guidelines were systematically applied to classify the pathogenicity of identified variants, incorporating multiple lines of evidence into a weighted framework: PVS1 (Pathogenic Very Strong): The novel variant identified in the ASH1L gene (c.2479A > T, p.Lys827Ter) results in a premature stop codon, predicted to lead to nonsense-mediated decay (NMD) and loss-of-function (LoF). This aligns with the established pathogenic mechanism of LoF for ASH1L-related disorders. PM2_Supporting (Pathogenic Moderate Supporting): The variant is absent in population databases, including ExAC and gnomAD, indicating it is a rare mutation. Following ACMG guidelines, the combined evidence supports classification of the ASH1L variant as “likely pathogenic.” The variant was confirmed by Sanger sequencing in the proband and family members, further validating its authenticity.
Genotype–phenotype correlationA review was conducted of all ASH1L truncating mutations (NM_018489.2) reported in HGMD through November 2024, with a summary of these mutations and their associated disease descriptions provided in Table 1. Truncating variants including frameshift and nonsense mutations cause significant protein malformations and typically lead to complete loss of function and haploinsufficiency.
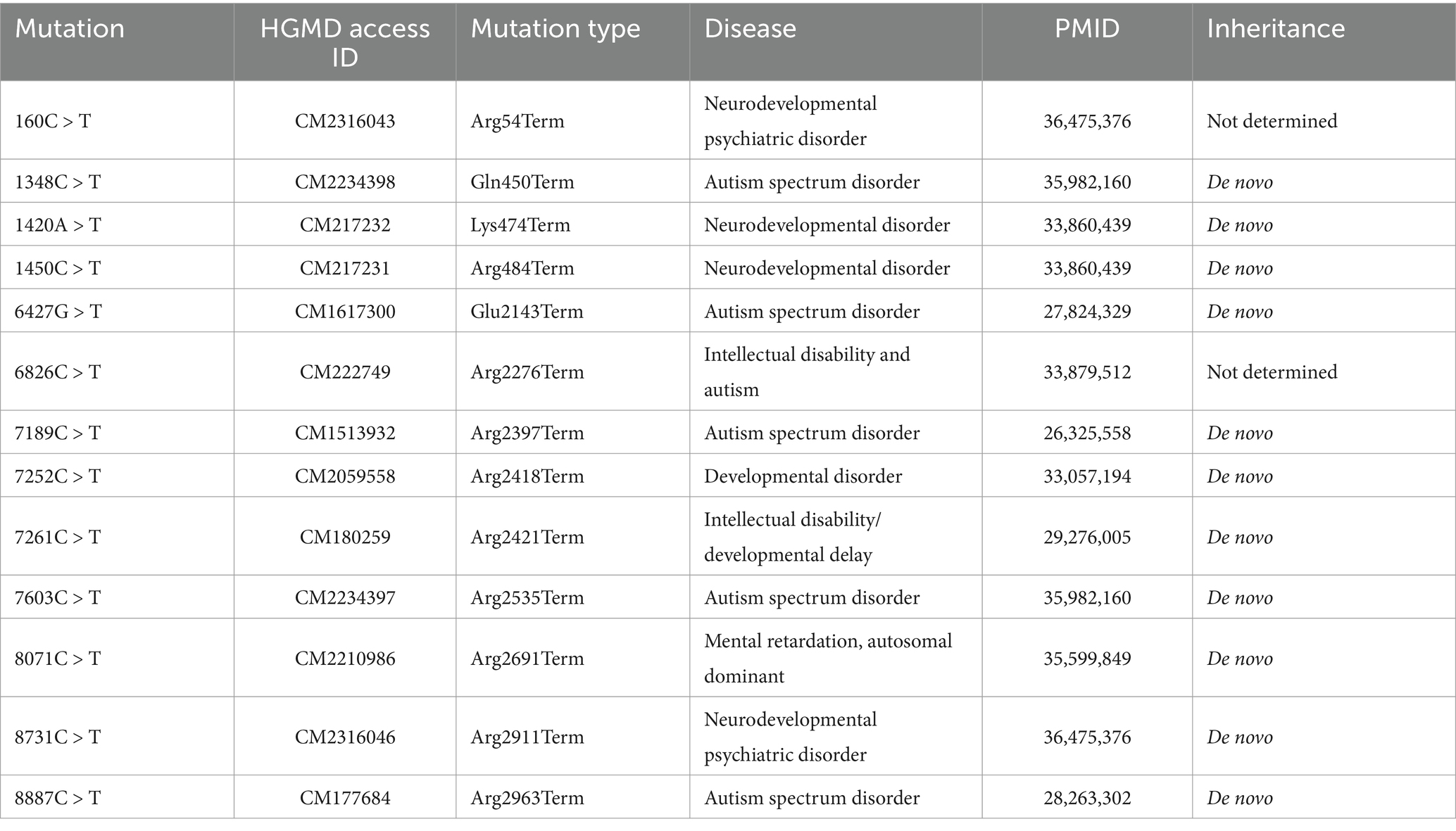
Table 1. Nonsense mutations described in the ASH1L gene, with their nucleotide position and associated phenotype.
Protein structure predictionThe protein sequence of ASH1L consisting of 2,965 amino acid residues was downloaded from uniprot web. By using the AlphaFold web server, the wild-type and mutant-type (ASH1L: c.2479A > T) 3D structure of the ASH1L protein could be predicted (25). Based on pLDDTs prediction scores, we selected the most reliable model for our studies, with higher scores indicating greater confidence in the structure. The prediction models were edited and visualized through PyMOL program.
Protein–protein interaction network and functional annotation analysisThe PPI network associated with ASH1L was constructed using STRING. The target genes in PPI network downloaded from STRING were used for reactome pathway enrichment analysis. The expression data was from the GTEX public database and was used to analyze the expression of ASH1L in different isoform and exon.
Results Identification of the novel heterozygous ASH1L nonsense variantThe patient’s whole-exome sequencing (WES) uncovered a novel heterozygous mutation (c.2479A > T/p.Lys827*) in the ASH1L gene. Cosegregation analysis revealed that the variant was passed down from the mother (Figure 1A). Sanger sequencing verified the presence or absence of this mutation in the family members (Figure 1B). Moreover, this variant have not been reported in any public population databases, including the 1,000 Genomes Project and GnomAD. The ASH1L c.2479A > T mutation is located on chromosome 1, specifically in exon 18 of the ASH1L gene. This mutation results in the substitution of adenine (A) with thymine (T) at nucleotide position 2,479, leading to the introduction of a premature stop codon at the protein level (p.Lys827*), which truncates the resulting protein (Figure 1C). The variant was conserved among species, including humans, rat, mice, cavpo, pig, bovine, and horse (Figure 1D). According to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpreting sequence variants, this mutation was classified as pathogenic, based on two key factors: (1) it is a predicted loss-of-function (LOF) variant in a gene where LOF is a known cause of disease. (2) No additional point mutations or copy number variants (CNVs) in candidate genes, including those linked to epilepsy, were detected under models of autosomal recessive.

Figure 1. A novo variant of ASH1L were identified in the patients. (A) The pedigrees and genotypes of the families are presented, with probands having undergone whole-exome sequencing (WES). Filled symbols represent affected individuals. (B) Sanger sequencing chromatograms display the ASH1L variant identified in the families. (C) Localization of the ASH1L: c.2479A > T variant found in the study. (D) Amino acid conservation of the novel variant p.Lys827* in different species.
Mutations in ASH1L carried by patient with ID affect the molecular structureIn order to analyze the effect of the novel variant NM_018489: c.2479A > T/p.Lys827*, on the structural integrity of the ASH1L protein, we employed the AlphaFold tool to predict potential changes in the protein’s structure. The NM_018489: c.2479A > T variant in ASH1L induces a frameshift mutation, resulting in a truncated ASH1L protein containing only 827 amino acids out of the 2,695 present in the full-length mature protein (Figures 2A,B). ASH1L is a multidomain protein composed of a long, unannotated N-terminus, a catalytic SET domain and three C-terminal histone binding domains: bromodomain (BRD), plant homeodomain (PHD) and bromo-associated homology (BAH) (26). The SET domain is responsible for histone methyltransferase (HMT) activity (16). The BRD domain of ASH1L targets acetylated chromatin to facilitating gene regulation. The BAH domain stabilizes ASH1L’s chromatin association and mediates interactions with other regulatory proteins (27). A crucial role of PHD domain is identifying histone modifications at specific sites and facilitating the recruitment of regulatory proteins (28). More and more research has reported that ASH1L mutations are associated with various neurodevelopmental disorders, including ID, ASD, and microcephaly (MCA) (Figure 2C) (19, 29, 30). The truncation caused by this variant results in the absence of key functional regions of ASH1L. Without these domains, the truncated ASH1L protein is likely unable to perform its typical biochemical functions. This alteration is expected to affect the stability of ASH1L, disrupt its enzymatic ability, and impede its role in chromatin remodeling, ultimately leading to disease pathology.
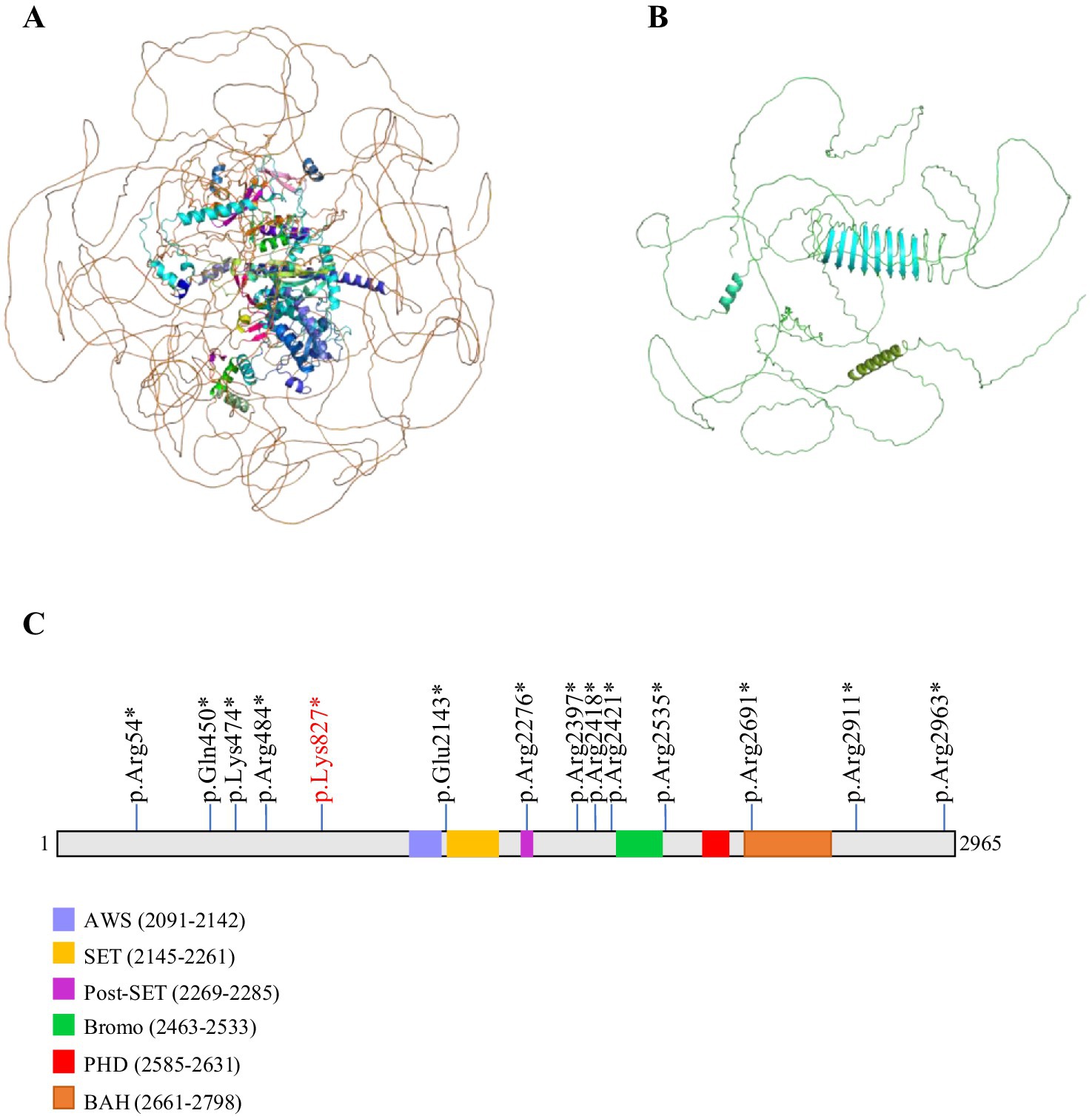
Figure 2. Potential impact of the c.2479A > T mutation on ASH1L protein structure. The wild-type structure of the ASH1L protein in (A) and the mutant protein structure shown in (B) contained a truncated ASH1L protein that contains 827 of the 2,695 amino acids of the mature protein. (C) Schematic diagram of ASH1L protein including five functional domains (AWS, SET, Post-SET, Bromo, PHD, BAH) are shown. All truncating mutations are indicated by sticks.
Functional effect of the heterozygous ASH1L nonsense variantTo identify gene-level characteristics that could support the biological basis for such a cluster, we began by searching the GTEx (Genotype-Tissue Expression) (see text footnote 6) and discovered that the full-length, canonical transcript is the only isoform expressed at an significant level in any adult human tissue (Figure 3A). In further support of this conclusion, exon-normalized expression was uniformly distributed across all exons in each tissue analyzed from the GTEx dataset (Figure 3B). Next, we investigated whether the existence of ASH1L have broader functional genomic effects. We then conducted a PPI network analysis using STRING to investigate potential genes that interact with ASH1L. As shown in Figure 3C, the PPI network consisted of 11 nodes. The top three proteins with higher degrees are MORF4L1, MORF4L1, and YY1AP1. The GO enrichment analysis (Figure 3D) showed that these genes were enriched in regulating methylate histone lysines, chromatin modifying enzymes and TGF dependent signaling in response to WNT. This suggests a multifaceted role for ASH1L in regulating chromatin structure and signal transduction processes.
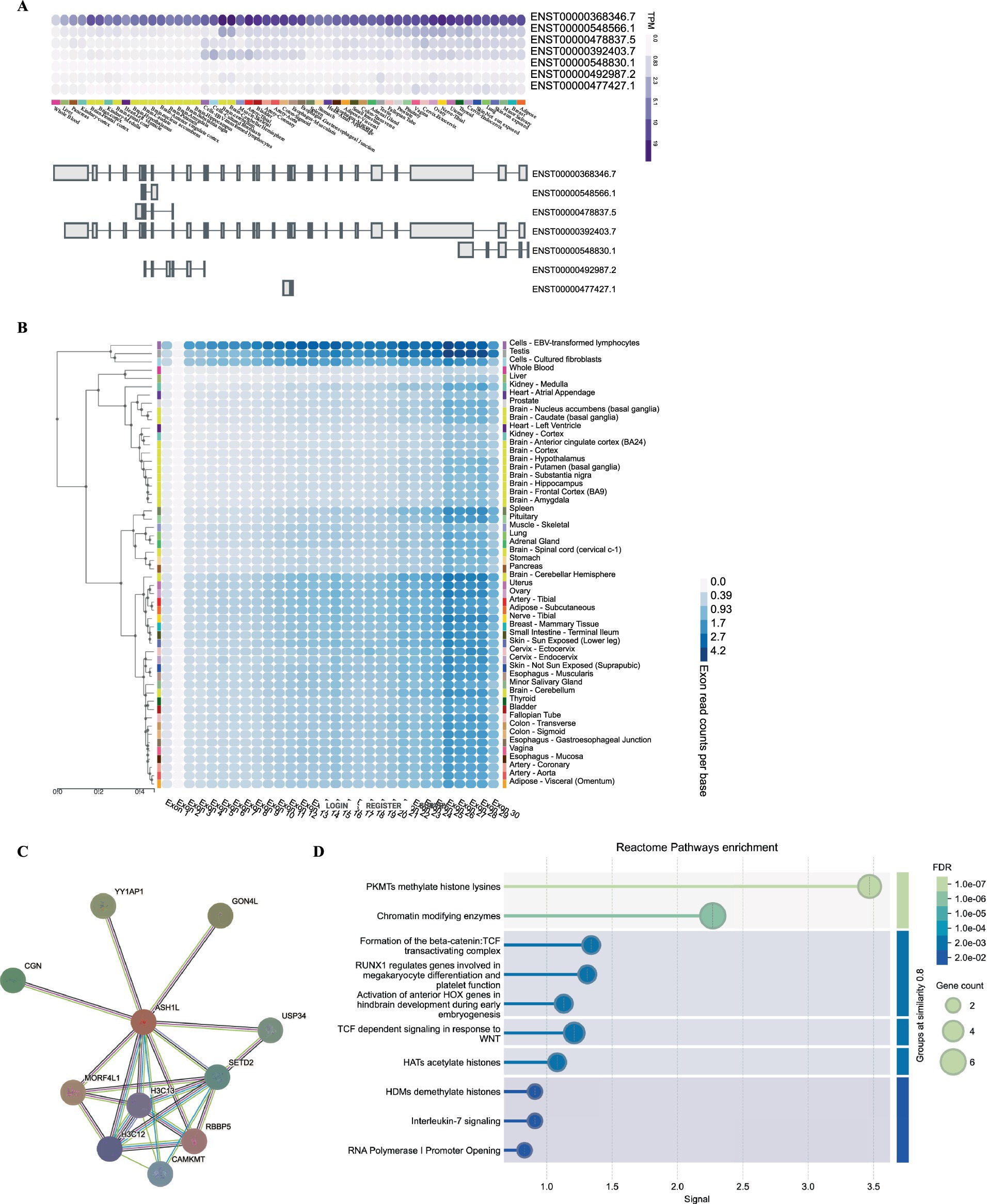
Figure 3. Gene expression and pathway analysis analysis of ASH1L. (A) GTEx data demonstrate that only a single, full-length ASHIL isoform (ENST00000368346.7) is expressed at an appreciable level in any adult human tissue represented. (B) The exon expression of ASH1L was obstained from GTEx data. (C) Bulk tissue gene expression for ASH1L. (D) The PPI network analysis of interacting genes with ASH1L and reactome pathway enrichment were downloaded from STRING.
DiscussionMild ID is a chronic neurodevelopmental disorder characterized by limitations in cognitive functioning and adaptive behaviors, with an increasing incidence observed in recent years (31). However, current drug treatments for patients with mild intellectual disabilities are often unsatisfactory and seriously affect their physical and mental health, particularly among youth. The primary interventions for managing mild ID include a variety of therapeutic approaches, such as educational support and behavioral therapies (32). Despite the advancements made in treatment strategies, the development of effective therapies for mild ID individuals remain a critical challenge. Therefore, improving our understanding of its pathogenesis would contribute to the development of new treatment in clinical.
The present study advances our understanding of ASH1L’s involvement in neurodevelopmental disorders by identifying a novel heterozygous nonsense variant, NM_018489.2: c.2479A > T (p.Lys827*), in a patient with mild ID. This discovery is aligned with the ASH1L’s critical role in chromatin remodeling and gene transcription within neural development pathways (33, 34). Given ASH1L’s role as a histone-lysine N-methyltransferase, this mutation likely disrupts crucial epigenetic processes necessary for normative brain development. This disruption broadens the phenotypic spectrum associated with ASH1L mutations and strengthens the link between ASH1L loss-of-function variants and a range of neurodevelopmental outcomes, including ID and ASD. It has been highlighted ASH1L play an important role in epigenetic regulation, particularly through its function in catalyzing the methylation of histone H3 at lysine 4 (H3K4) (35). H3K4me3 promotes gene activation when NURF complexes are present. It maintains chromatin in an active “on” state through the plant homeodomain (PHD) domain. This allows transcription factors to access DNA within the chromatin (36). ASH1L regulates the expression of essential developmental genes by antagonizing polycomb- mediated gene silencing. This action finally limits the accessibility of target genes.
As seen in other neurodevelopmental disorders, H3K4 dysregulation can lead to varied neurobehavioral and cognitive phenotypes depending on the specific mutation and affected neural pathways (37–39). Moreover, previous research has established a phenotypic range in ASH1L-associated conditions with pathogenic variants linked to severe and mild cognitive impairments, language deficits, and neurobehavioral disturbances (40, 41). The presence of a novel nonsense variant, p.Lys827*, adds to this evidence, emphasizing the variability in clinical outcomes associated with ASH1L mutations. Mouse models deficient in ASH1L have displayed abnormal cortical neuron differentiation and craniofacial abnormalities, supporting a strong genotype–phenotype correlation and mirroring human neurodevelopmental features associated with ASH1L loss-of-function (42, 43). However, most human studies to date have focused on more severe clinical cases of ASH1L-associated ID. This makes our discovery of a nonsense variant in a mildly affected individual particularly relevant for expanding the clinical profile of ASH1L-related neurodevelopmental disorders.
Importantly, our findings in nonsense mutation demonstrates WES’s value in identifying rare variants and establishes ASH1L as a key candidate for further study in mild ID. A limitation of our study was that WES was performed only on the proband sample. This single-sample approach restricts our ability to fully assess the inheritance pattern and the broader genetic context of the mutation. To address this limitation, future research should include family-based sequencing to better understand how the mutation is inherited and its potential interaction with other genetic factors, providing a more comprehensive view of ASH1L-related disorders. Future studies could leverage multi-omics approaches to explore the transcriptional, proteomic, and epigenetic alterations resulting from ASH1L loss-of-function mutations, particularly nonsense variants. Transcriptomic analyses could provide insights into specific gene expression disruptions linked to ASH1L mutations, while proteomic studies might elucidate downstream signaling pathways that are dysregulated. However, there are significant methodological differences between the construction, tissue sampling, RNA preparation, and analysis of the two models. Moreover, the use of patient-derived induced pluripotent stem cells (iPSCs) and neuronal differentiation models could enable in-depth functional studies, helping to identify molecular targets for potential therapeutic interventions. Additionally, to understand how ASH1L haploinsufficiency affects cognitive and behavioral phenotypes, it would help to do long-term studies on groups of people and use brain imaging to see how specific brain areas change in structure and function over time.
In conclusion, the identification of the novel ASH1L p.Lys827* nonsense variant contributes significant insights to the complex genetic landscape of ID and ASH1L-related neurodevelopmental disorders. By broadening the clinical spectrum associated with ASH1L mutations, this study provides a foundation for genotype–phenotype correlations and emphasizes the necessity for comprehensive genetic and functional analyses in mild ID cases. Future research should continue to investigate the specific pathways affected by ASH1L mutations and explore potential targeted therapeutic interventions that address the potential epigenetic dysregulation in ASH1L-related disorders.
Data availability statementThe raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statementThe studies involving humans were approved by the Ethics Committee of the Ganzhou Maternal and Child Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributionsBL: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. WX: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. SH: Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Writing – original draft, Writing – review & editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research is supported by the Ganzhou city science and technology plan project (GZ2024YLJ203).
AcknowledgmentsThe patient and their family are greatly appreciated for their invaluable participation in this study. We thank Yarong Tian from the University of Gothenburg for helpful discussions.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes References1. Ilyas, M, Mir, A, Efthymiou, S, and Houlden, H. The genetics of intellectual disability: advancing technology and gene editing. F1000Res. (2020) 9:F1000FacultyRev-22. doi: 10.12688/f1000research.16315.1
PubMed Abstract | Crossref Full Text | Google Scholar
2. Schalock, RL, Luckasson, RA, Shogren, KA, Borthwick-Duffy, S, Bradley, V, Buntinx, WH, et al. The renaming of mental retardation: understanding the change to the term intellectual disability. Intellect Dev Disabil. (2007) 45:116–24. doi: 10.1352/1934-9556(2007)45[116:TROMRU]2.0.CO;2
PubMed Abstract | Crossref Full Text | Google Scholar
4. Wehmeyer, ML, Buntinx, WH, Lachapelle, Y, Luckasson, RA, Schalock, RL, Verdugo, MA, et al. The intellectual disability construct and its relation to human functioning. Intellect Dev Disabil. (2008) 46:311–8. doi: 10.1352/1934-9556(2008)46[311:TIDCAI]2.0.CO;2
PubMed Abstract | Crossref Full Text | Google Scholar
5. Shevell, M. Global developmental delay and mental retardation or intellectual disability: conceptualization, evaluation, and etiology. Pediatr Clin N Am. (2008) 55:1071–84. doi: 10.1016/j.pcl.2008.07.010
PubMed Abstract | Crossref Full Text | Google Scholar
9. Rauch, A, Hoyer, J, Guth, S, Zweier, C, Kraus, C, Becker, C, et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A. (2006) 140A:2063–74. doi: 10.1002/ajmg.a.31416
PubMed Abstract | Crossref Full Text | Google Scholar
10. Patel, DR, Cabral, MD, Ho, A, and Merrick, J. A clinical primer on intellectual disability. Transl Pediatr. (2020) 9:S23–35. doi: 10.21037/tp.2020.02.02.
Crossref Full Text | Google Scholar
11. McFayden, TC, Culbertson, S, DeRamus, M, Kramer, C, Roush, J, and Mankowski, J. Assessing autism in deaf/hard-of-hearing youths: interdisciplinary teams, COVID considerations, and future directions. Perspect Psychol Sci. (2023) 18:1492–507. doi: 10.1177/17456916231178711
PubMed Abstract | Crossref Full Text | Google Scholar
13. di, E, Riberi, E, Belligni, EF, Biamino, E, Spielmann, M, Ala, U, et al. Copy number variants analysis in a cohort of isolated and syndromic developmental delay/intellectual disability reveals novel genomic disorders, position effects and candidate disease genes. Clin Genet. (2017) 92:415–22. doi: 10.1111/cge.13009
PubMed Abstract | Crossref Full Text | Google Scholar
14. Harripaul, R, Noor, A, Ayub, M, and Vincent, JB. The use of next-generation sequencing for research and diagnostics for intellectual disability. Cold Spring Harb Perspect Med. (2017) 7:a026864. doi: 10.1101/cshperspect.a026864
PubMed Abstract | Crossref Full Text | Google Scholar
15. Zhao, X, Lin, S, Ren, H, Sun, S, Zheng, L, Chen, LF, et al. The histone methyltransferase ASH1L protects against bone loss by inhibiting osteoclastogenesis. Cell Death Differ. (2024) 31:605–17. doi: 10.1038/s41418-024-01274-w
PubMed Abstract | Crossref Full Text | Google Scholar
16. Zhang, C, Xu, L, Zheng, X, Liu, S, and Che, F. Role of Ash1l in Tourette syndrome and other neurodevelopmental disorders. Dev Neurobiol. (2021) 81:79–91. doi: 10.1002/dneu.22795
PubMed Abstract | Crossref Full Text | Google Scholar
17. Zhu, L, Li, Q, Wong, SH, Huang, M, Klein, BJ, Shen, J, et al. ASH1L links histone H3 lysine 36 Dimethylation to MLL leukemia. Cancer Discov. (2016) 6:770–83. doi: 10.1158/2159-8290.CD-16-0058
PubMed Abstract | Crossref Full Text | Google Scholar
18. De Ligt, J, Willemsen, MH, van Bon, BW, Kleefstra, T, Yntema, HG, Kroes, T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. (2012) 367:1921–9. doi: 10.1056/NEJMoa1206524
PubMed Abstract | Crossref Full Text | Google Scholar
19. Okamoto, N, Miya, F, Tsunoda, T, Kato, M, Saitoh, S, Yamasaki, M, et al. Novel MCA/ID syndrome with ASH1L mutation. Am J Med Genet A. (2017) 173:1644–8. doi: 10.1002/ajmg.a.38193
PubMed Abstract | Crossref Full Text | Google Scholar
20. Wang, T, Guo, H, Xiong, B, Stessman, HA, Wu, H, Coe, BP, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. (2016) 7:13316. doi: 10.1038/ncomms13316
PubMed Abstract | Crossref Full Text | Google Scholar
21. Andjelkovic, M, Klaassen, K, Skakic, A, Marjanovic, I, Kravljanac, R, Djordjevic, M, et al. Characterization of 13 novel genetic variants in genes associated with epilepsy: implications for targeted therapeutic strategies. Mol Diagn Ther. (2024) 28:645–63. doi: 10.1007/s40291-024-00720-2
PubMed Abstract | Crossref Full Text | Google Scholar
22. Zhang, C, Liu, W, Xu, L, Liu, S, and Che, F. Abnormal H3K4 enzyme catalytic activity and neuronal morphology caused by ASH1L mutations in individuals with Tourette syndrome. Eur Child Adolesc Psychiatry. (2024) 33:3913–23. doi: 10.1007/s00787-024-02437-3
PubMed Abstract | Crossref Full Text | Google Scholar
23. Zhang, XY, and Li, Y. PHD-BAH domain in could recognize H3K4 methylation and regulate the malignant behavior of Cholangio carcinoma. Anti Cancer Agent Med Chem. (2024) 24:1264–74. doi: 10.2174/0118715206312004240712072532
PubMed Abstract | Crossref Full Text | Google Scholar
24. Toolan, KP, McGrath, BT, Brinkmeier, ML, Camper, SA, and Bielas, SL. Ash1 l loss-of-function results in structural birth defects and altered cortical development. Brain. (2024) 148:55–68. doi: 10.1093/brain/awae218
PubMed Abstract | Crossref Full Text | Google Scholar
25. Abramson, J, Adler, J, Dunger, J, Evans, R, Green, T, Pritzel, A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. (2024) 630:493–500. doi: 10.1038/s41586-024-07487-w
PubMed Abstract | Crossref Full Text | Google Scholar
26. An, S, Yeo, KJ, Jeon, YH, and Song, JJ. Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. J Biol Chem. (2011) 286:8369–74. doi: 10.1074/jbc.M110.203380
PubMed Abstract | Crossref Full Text | Google Scholar
27. Steffen, PA, Altmutter, C, Dworschak, E, Junttila, S, Gyenesei, A, Zhu, X, et al. The Trithorax group protein ASH1 requires a combination of BAH domain and AT hooks, but not the SET domain, for mitotic chromatin binding and survival. Chromosoma. (2021) 130:215–34. doi: 10.1007/s00412-021-00762-z
PubMed Abstract | Crossref Full Text | Google Scholar
28. Rogawski, DS, Ndoj, J, Cho, HJ, Maillard, I, Grembecka, J, and Cierpicki, T. Two loops undergoing concerted dynamics regulate the activity of the ASH1L histone methyltransferase. Biochemistry. (2015) 54:5401–13. doi: 10.1021/acs.biochem.5b00697
PubMed Abstract | Crossref Full Text | Google Scholar
29. Liu, W, Xu, L, Zhang, C, Shen, L, Dong, J, Zhang, H, et al. ASH1L may contribute to the risk of Tourette syndrome: combination of family-based analysis and case-control study. Brain Behav. (2022) 12:e2539. doi: 10.1002/brb3.2539
PubMed Abstract | Crossref Full Text | Google Scholar
30. Shen, W, Krautscheid, P, Rutz, AM, Bayrak-Toydemir, P, and Dugan, SL. De novo loss-of-function variants of ASH1L are associated with an emergent neurodevelopmental disorder. Eur J Med Genet. (2019) 62:55–60. doi: 10.1016/j.ejmg.2018.05.003
PubMed Abstract | Crossref Full Text | Google Scholar
32. Osugo, M, and Cooper, SA. Interventions for adults with mild intellectual disabilities and mental ill-health: a systematic review. J Intellect Disabil Res. (2016) 60:615–22. doi: 10.1111/jir.12285
PubMed Abstract | Crossref Full Text | Google Scholar
33. Cheon, S, Culver, AM, Bagnell, AM, Ritchie, FD, Vacharasin, JM, McCord, MM, et al. Counteracting epigenetic mechanisms regulate the structural development of neuronal circuitry in human neurons. Mol Psychiatry. (2022) 27:2291–303. doi: 10.1038/s41380-022-01474-1
PubMed Abstract | Crossref Full Text | Google Scholar
34. Gregory, GD, Vakoc, CR, Rozovskaia, T, Zheng, X, Patel, S, Nakamura, T, et al. Mammalian ASH1L is a histone methyl transferase that occupies the transcribed region of active genes. Mol Cell Biol. (2007) 27:8466–79. doi: 10.1128/Mcb.00993-07
PubMed Abstract | Crossref Full Text | Google Scholar
35. Yu, M, Jia, Y, Ma, Z, Ji, D, Wang, C, Liang, Y, et al. Structural insight into ASH1L PHD finger recognizing methylated histone H3K4 and promoting cell growth in prostate cancer. Front Oncol. (2022) 12:906807. doi: 10.3389/fonc.2022.906807
PubMed Abstract | Crossref Full Text | Google Scholar
36. Wysocka, J, Swigut, T, Xiao, H, Milne, TA, Kwon, SY, Landry, J, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. (2006) 442:86–90. doi: 10.1038/nature04815
PubMed Abstract | Crossref Full Text | Google Scholar
37. Collins, BE, Greer, CB, Coleman, BC, and Sweatt, JD. Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics Chromatin. (2019) 12:7. doi: 10.1186/s13072-018-0251-8
PubMed Abstract | Crossref Full Text | Google Scholar
38. Wang, S, Bleeck, A, Nadif, N, Kleefstra, T, van, J, and Schubert, D. SETD1A mediated H3K4 methylation and its role in neurodevelopmental and neuropsychiatric disorders. Front Mol Neurosci. (2021) 14:772000. doi: 10.3389/fnmol.2021.772000
PubMed Abstract | Crossref Full Text | Google Scholar
40. Krumm, N, Turner, TN, Baker, C, Vives, L, Mohajeri, K, Witherspoon, K, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. (2015) 47:582–8. doi: 10.1038/ng.3303
留言 (0)