
記住我
Leukemias are a group of hematological cancers characterized by the abnormal proliferation of partially-differentiated hematopoietic stem cells (HSCs) (1). Although there are multiple types of leukemia, the four most common are acute and chronic myeloid leukemias (AML and CML) and acute and chronic lymphocytic leukemias (ALL and CLL). Data from the National Cancer Institute (NCI) indicate that the five-year survival rate for patients is 70.6% for CML, 71.3% for ALL, and 88% for CLL. This contrasts with AML, where it is only 31.9%. AML survival negatively correlates with patient age and the presence of comorbidities; unsurprisingly, both conditions also reduce treatment options. AML is also characterized by particularly aggressive progression; if left untreated, patients can succumb to disease within weeks.
As can be inferred from their names, different blast cell populations are observed in different leukemias. AML shows excess myeloblast proliferation, which compromises normal signaling and differentiation, resulting in the accumulation of an immature and poorly functioning population of cells. This compromises normal hematopoiesis, leading to anemia and immune dysfunction. This is demonstrated by the susceptibility to opportunistic diseases (e.g., bacterial infections) in AML patients. Ultimately, AML patients succumb to some combination of leukostasis (a condition characterized by extremely elevated blast count and decreased oxygen flow to tissues, possibly due to blasts preventing erythrocyte flow), organ failure, and/or infection by common pathogens that are normally cleared by the immune system.
In most cases, AML blasts exhibit profound metabolic and mitochondrial changes that promote unrestrained cell division (discussed further in Section 2). The metabolic reprogramming and mitochondrial dysfunction seen in AML sensitize the cells to mitochondrial damage, which has recently sparked considerable interest in targeting mitochondria for therapeutic effect (2–5). For example, drugs that disrupt mitochondrial homeostasis by inducing or inhibiting mitophagy (autophagic degradation of mitochondria) are one area of particular interest (discussed further in Section 3) (6, 7).
The current standard first-line treatment regimen for AML is called induction and consolidation therapy. Its purpose is to reduce the number of leukemic blast cells in the body (de-bulk). During induction, patients are treated with a high dose of the cytosine nucleoside analog cytarabine (araC) in combination with an anthracycline (such as daunorubicin, doxorubicin, or idarubicin) that intercalates into DNA to disrupt topoisomerase II function. Together, these drugs induce profound cell death in proliferating myeloblasts in the bone marrow and blood (8). If induction is successful, patients are brought to a state called remission, clinically defined as less than 5% circulating leukemic blasts and recovery of normal hematopoiesis (8). De-bulking the leukemic blast cells often allows the body to temporarily regain hematopoietic homeostasis.
Once remission is achieved, induction is followed by consolidation. During this stage, patients are usually treated with a high dose of araC to prevent further myeloblast proliferation (or, preferably, to further reduce the number of leukemic cells). In some cases, consolidation is not done with araC but with other drugs selected to target specific AML mutations (1, 8, 9). Without consolidation, patients often experience rapid relapse because induction is ineffective against slowly cycling leukemic stem cells (LSCs), which co-exist in the bone marrow niche with their leukemic blast progeny and normal HSCs (10). Interestingly, available evidence indicates that LSCs are derived from partially differentiated HSCs that have aberrantly regained self-renewal capacity (10). LSCs that have not succumbed to treatment resume producing rapidly-cycling myeloblasts, promptly restoring the leukemic state.
Irradiation is another AML treatment, often used following induction and consolidation, to destroy bone marrow and nucleated blood cells, whether cancerous or not (11–13). Once these cell populations have been removed, an allogenic hematopoietic stem cell transplant (allo-HSCT) is performed to provide the patient with a functional population of bone marrow cells (8). Patients commonly receive maintenance chemotherapy after allo-HSCT, particularly those with a high risk of relapse. In some cases, this aggressive course of treatment is sufficient to abolish the leukemic cells and restore patients to long-term health. In many cases, however, this aggressive treatment leads to leukemia cells that are resistant to the treatments being used (11).
Relapsed and refractory AML, which are indicative of treatment failure, are primarily caused by drug resistance. The emergence of treatment-resistant clonal lines in patients is another cause for low overall AML patient survival. There are multiple mechanisms of cancer drug resistance, including aberrant expression of drug-resistance proteins and microRNAs, dysregulation of signaling pathways, and often the same genetic alterations that caused the illness in the first place (14–17). Many of these dysregulations implicate mitochondrial signaling and metabolic pathways. Other resistance mechanisms include modifying or preventing incorporation of the drug. For example, mutations in deoxycytidine kinase (dCk) activity reduce conversion of araC to a dideoxy form that can be incorporated into replicating DNA. AraC can also be converted into an alternate, unusable metabolite to prevent incorporation (18–20).
Resistance in AML is often driven by decreased import and increased efflux (21). Alterations to import and efflux are common mechanisms of resistance observed for almost all FDA-approved treatments, including anthracyclines, mitoxantrone, etoposide, and methotrexate (22–26). For example, increased expression of the drug efflux pump, P-glycoprotein (Pgp/ABCB1), is associated with poorer prognosis (27).
Mutations in surface antigens can also confer resistance. For example, CD33 is a myeloid marker expressed in about 90% of AML patient cells, but rarely expressed on normal HSCs (28). The antibody-drug conjugate gemtuzumab ozogamicin (GO), which is comprised of an anti-CD33 monoclonal antibody fused to the antitumor antibiotic calicheamicin, shows resistance linked to the activity of glycogen synthase kinase-3 (GSK3). The effectiveness of GO decreases with increased activity of GSK3. Such resistance has been overcome with parallel use of GSK3 inhibitors in cell lines (29). Although GSK3 is tightly linked to a wide variety of mitochondrial functions, even in cancer cells, GSK3-mediated resistance to GO was attributed to reduced CD33 expression (reducing the efficacy of antibody targeting), increased lysosomal drug degradation, increased export, and increased expression of the anti-apoptotic protein BCL-2 (29, 30).
In still other cases, resistance to therapies is caused by further changes to mitochondrial metabolism that reduce treatment effectiveness. Examples of this include upregulating the anti-apoptotic protein BCL-2, upregulating amino acid catabolism, and utilizing alternative energy pathways such as lipid oxidation (31–33). This review will discuss recent findings and progress in leukemia treatment, particularly focusing on the importance of targeting the mitochondria using changes in mitophagy to bolster AML treatment.
2 Metabolic reprogramming in AML2.1 Changes in oxidative phosphorylationMost AML cells have mutations in one or more genes that affect cell growth or metabolic function and anomalies in mitochondrial metabolism are frequently observed (3, 4, 34–37). Like all cancer cells, leukemic cells need to increase their energy production to support their increased cell division (5, 38). For many cancer types, this drive for increased energy induces so-called Warburg metabolism, which involves upregulation of glycolysis and reduced OXPHOS (39). Contrastingly, AML blasts instead appear to show an increase in dependency on OXPHOS compared to most other cancer types (2).
One common cause of the increased OXPHOS in AML is mutation of the membrane-bound receptor Fms-like tyrosine kinase 3 (FLT3). Normal FLT3 activity is associated with regular hematological cell growth and development (40). But internal tandem duplications of the kinase domain (known as FLT3-ITD, found in about 20% of AML cases) and point mutations or deletions in the kinase domain (FLT3-TKD, present in about 10% of AML cases) have been shown to disrupt Ras signaling and upregulate expression of PDP1, a key regulator of the pyruvate dehydrogenase complex (41, 42). This increases pyruvate flux through the citrate cycle, increasing OXPHOS activity, energy production, and ROS generation.
Several inhibitors of FLT3, such as midostaurin, gilteritinib, quizartinib, or sorafenib, have been identified and tested for use in AML patients with FLT3 mutations (43). Unfortunately, FLT3 inhibitor monotherapy is frequently unsuccessful and often results in relapse and resistance (44). Combining various FLT3 inhibitors with commercial chemotherapeutics has been attempted in clinical trials, with mixed results (45). Notably, the addition of midostaurin to induction and consolidation treatment demonstrated an overall survival benefit in the clinical trial, RATIFY, and was approved by the FDA (9, 46–48). FLT3 inhibitors have had heterogenous success as maintenance therapy after allo-HSCT, thus further investigation into relapse-free cases is warranted (9, 47, 49–51).
Mutations in isocitrate dehydrogenase (IDH, which are found in about 20% of AML patients) are associated with increased mitochondrial oxidative metabolism, including Complex I activity, respiration, and fatty acid oxidation. IDH inhibitors, such as ivosidenib or enasidenib, were evaluated and showed patterns similar to FLT3 inhibitors: monotherapy often showed temporary clinical utility, but was followed by the rapid rise of resistance (52–54). Combination with other therapies was roughly twice as effective, but relapse and resistance were still common, making patients with mutations in IDH a key population that urgently needs useful therapies. Disruptions of IDH and FLT3 not only support AML cell survival, they can also promote resistance to chemotherapy (55–57).
Although AML cells consume more oxygen at the basal metabolic level, the efficiency of ATP generation does not improve because the cells exhibit significant proton leak and poor coupling efficiency (ratio of oxygen consumed for ATP production to oxygen lost in proton leak) (36). OXPHOS is driven by the proton gradient generated by the electron transport chain (ETC) pumping protons from the mitochondrial matrix to the intermembrane space. Some protons pass back across the mitochondrial membrane without contributing towards ATP production (i.e., leak) (58–61). OXPHOS dependency increases the level of reactive oxygen species (ROS) making AML cells more sensitive to ROS levels (61). AML blasts also have reduced spare reserve capacity, which is the difference between their basal metabolic load and their maximum metabolic rate. This increase in OXPHOS drives parallel increases in ROS production and accumulation of mitochondrial damage, which is likely a cause for the observations that AML blasts have a reduced capacity to tolerate exogenous oxidative stress (34, 35). For example, while studying responses of the NCI-60 cell panel to a variety of drugs, we observed that leukemic cells show particular sensitivity to mitochondrial damage (3). Further analysis demonstrated that, although they have nearly twice as many mitochondria as healthy hematopoietic cells, AML cells have poor respiration capacity and coupling efficiency. This also led us to believe that mitochondrial damage may be a viable adjuvant to other chemotherapies.
These mitochondrial changes are likely one reason that AML blasts show increased mitochondrial mass and increased rates of mitochondrial biogenesis (34). Other reports have shown that blasts also exhibit a greater dependence upon fatty acid oxidation (37), particularly very long-chain fatty acid oxidation (62). Alteration of sphingolipid composition, specifically reduction of ceramide, also leads to increased mitochondrial biogenesis and OXPHOS (63). This mitochondrial remodeling was found to contribute to AML cells’ resistance to the combination of daunorubicin and araC, and standalone doxorubicin treatment (63, 64). Similarly, these cells are sensitive to treatment with ceramides, particularly when combined with an OXPHOS inhibitor (35). Upon treatment with the combination of C6-ceramide and tamoxifen, mitochondria were seen to colocalize with autophagosomes, indicating activation of autophagy and mitophagy (65). Remarkably, exposure to either drug alone had no apparent effect on AML cell viability, but the combination resulted in a strong additive effect. One possible explanation for this outcome is that the mitochondrial damage that accumulates during AML overtaxes mitochondrial recycling pathways, and further stimulation tips the cell into activation of programmed cell death pathways. This supports the idea that mitophagy may be used to eliminate treatment-resistant AML cells (see Section 3).
Although it is well-recognized that OXPHOS activity increases ROS production, surprisingly, inhibiting OXPHOS also induces the production of ROS in AML cells (66, 67). For example, treatment of OCI-AML3 cells with the OXPHOS inhibitor IACS-010759 alone, or in combination with low-dose araC, significantly increased ROS production (67). Increased ROS levels after treatment with IACS-010759 and araC also trigger mitochondrial fission that leads to mitophagy. This combination of mitochondrial activities induces the resistance of AML to OXPHOS inhibition by promoting mitochondrial transfer from mesenchymal cells and, unfortunately, renders this combination therapy relatively ineffective (67). Nevertheless, OXPHOS inhibitors and other mitochondria-targeting chemotherapeutics are widely used to improve treatment response (68).
2.2 Increased glutamine utilization in AMLAnother significant example of metabolic reprogramming in AML cells is the highly increased use of glutaminolysis by blasts and LSCs, so that they are occasionally referred to as glutamine-addicted leukemia (69–72). AML cells rely on glutamine as a carbon source for energy production and its derivative, glutathione, for controlling ROS. Glutamine is transported into the cell, broken down into glutamate, and oxidized into α-ketoglutarate before entering the citric acid cycle (CAC) to produce energy and bypass the need for glucose in energy production (73). Additionally, the direct utilization of α-ketoglutarate bypasses the rate-limiting step of CAC: converting isocitrate into α-ketoglutarate (74). The products of the CAC are used to drive OXPHOS by activating the complexes of the ETC to create the proton gradient for ATP synthesis. The utilization of glutamine over glucose increases energy production via OXPHOS, making glutaminolysis a promising therapeutic target.
Drugs targeting glutamine uptake, glutamine antagonists, glutaminase inhibitors, and asparaginases are all under investigation (6). Targeting glutaminolysis decreases OXPHOS capacity and inhibits the CAC cycle. However, these compounds are also subject to resistance. Leukemic cells can be rescued by excess CAC intermediates derived from glutaminolysis, and acquire chemoresistance through metabolic shifts to favor other carbon sources such as glucose (75, 76). Additionally, immune cells rely on glutaminolysis to generate excess energy, resulting in toxic side effects in some cases (77). Therefore, additional characterization of glutaminolysis is needed to generate more specific and less toxic metabolism-targeting compounds.
Glutamine metabolism also provides building blocks for glutathione, which reduces cellular ROS (78). Inhibiting the enzymes responsible for converting oxidized glutathione (GSSG) to its reduced form (GSH) increased ROS levels and killed cells (79, 80). GSH is therefore also a potential therapeutic target (81).
2.3 Additional metabolic reprogramming in LSCs and the bone marrow microenvironmentLSCs also exhibit considerable metabolic reprogramming. These cells co-exist in the bone marrow microenvironment with blast cells and more typical HSCs, but LSCs are thought to be differentiated from normal HSCs by the heterogenous expression of a variety of surface markers, including CD32, CD44, CD45RA, CD47, CD96, CD123, and TIM3 (10, 82). Unlike HSCs, which divide and differentiate at a much slower rate and can rely on glycolysis and the limited OXPHOS possible in the bone marrow niche for their energy needs (10), LSCs divide more rapidly, generating a higher energetic demand compared to HSCs. LSCs fulfill this need for energy by increasing OXPHOS and mitochondrial mass, like AML and CML blasts (10, 82, 83). As the bone marrow microenvironment is comparatively hypoxic, LSCs compensate this condition by increasing lipid oxidation and glutaminolysis to support this requirement (10, 32, 82). AML blast cells and LSCs cultured under hypoxic conditions have been shown to upregulate both general macroautophagy and mitophagy to support development and mitigate cellular damage (84, 85). Activation of these pathways helps to prevent build-up of ROS that may induce apoptosis and is likely to be a key factor in the low levels of ROS observed in LSCs (86).
The bone marrow microenvironment has also been shown to protect LSCs and leukemic blast cells from chemotherapeutics. Bone marrow stromal cells transfer mitochondria to AML blasts via leukemia-derived tunneling nanotubes, promoting chemoresistance (66, 67). A study involving a niche-like coculture system showed that up to 14% of the mitochondria in stromal cells are transferred to AML cells with which they are in physical contact, leading to a 50% increase in mitochondrial ATP production (87). Organelle transfer is also stimulated by superoxide production by AML-derived NADPH oxidases (NOX2), which increase ROS in the bone marrow stromal cells (66, 67). Transferred mitochondria from bone marrow stromal cells not only contributes to ATP generation via the CAC and OXPHOS, but also to production of GSH, which is likely to ameliorate the ROS and help limit oxidative crisis (88).
LSCs also exhibit an increase in mitochondrial number but a reduction in size due to increased rates of mitochondrial fission (86, 89). The AMPK/FIS1 signaling pathway is constitutively expressed in AML LSCs, promoting fission, activating mitophagy, and sustaining proliferation. This activity has also been theorized to assist in promoting stemness in the LSCs (86, 89). Consistent with this, loss of FIS1 expression leads to upregulation of hematopoietic and myeloid differentiation markers (86). A similar phenotype is also observed after loss of mitophagic regulators (PINK1, TBC1D15, etc.), indicating that mitochondrial fission and mitophagy are critical for the maintenance of LSC viability (86).
3 Mitophagy in AML3.1 Mechanisms of mitochondrial autophagic degradationAutophagy, or self-eating, is a pro-survival mechanism that allows cells to recycle large portions of their machinery, ranging from portions of the cytoplasm and its protein components to whole organelles. Cellular material is engulfed into a membrane-bound compartment called an autophagosome that then fuses with lysosomes to enable the degradation of the cellular materials. Recycling of organelles and other cellular structures decomposes them into their constituents, which can then be used to replenish necessary metabolites or to generate new organelles (86). Autophagy is often triggered by changes in cellular environment, particularly nutrient deprivation, oxidative stress, or other stress signals. Autophagy is also frequently upregulated in cancer cells, including AML, where it is thought to provide raw materials (e.g., lipids and amino acids) to support cell division (90–93).
Autophagic degradation is also used as a selective mechanism for recycling damaged or dysfunctional mitochondria (known as mitophagy). Mitochondria absorb damage from a variety of causes, including external, stress-inducing environmental stimuli (e.g., carcinogens, pesticides, and heavy metals) and the internal metabolic processes that produce ATP (94–97). These factors damage the proteins, lipids, and DNA of the organelle, reducing functionality. Worse, this damage often decreases OXPHOS efficiency, further increasing ROS levels in a vicious, downward cycle. Mitochondria damaged in this way are tagged by pro-mitophagic proteins that facilitate autophagosome recruitment and mitochondrial recycling (see below). Cells have relatively few mitochondrial repair pathways; therefore, they heavily rely on mitophagic turnover for resolving damage (98, 99).
Two main mechanisms of mitophagy have been identified in mammals (Figure 1), and both have been linked to carcinogenesis and have been implicated in the progression and poor prognosis of human AML (100, 101). The first mechanism is regulated by the well-known PTEN1-induced kinase (PINK1)-Parkin pathway, which has been subjected to more study than the other known pathways. Under normal circumstances, PINK1 is localized to the outer membrane of mitochondria. Normal mitochondrial import processes bring the protein into the organelle, where it is cleaved by PARL, a protease resident in the mitochondrial intermembrane space. The remaining portion of PINK1 returns to the cytosol where it is degraded by a proteasome (102). If mitochondrial membrane potential is lost (i.e., the mitochondrial membrane is depolarized) or protein import is impaired for some other reason, PINK1 accumulates on the outer mitochondrial membrane. Under these conditions, PINK1 is auto- and transphosphorylated, licensing the phosphorylation of additional targets, such as the ubiquitin ligase Parkin (Figure 1). Once activated, Parkin catalyzes the transfer of ubiquitin to various mitochondria-resident transmembrane proteins, allowing them to be recognized by autophagosomal sequestration machinery, such as the protein MAP1-LC3 (103, 104). Ubiquitination of outer mitochondrial membrane proteins facilitates their recognition by additional receptors, such as NIX (NIP3-like protein X), BNIP-3 (BCL-2/adenovirus E1B 19 kDa interacting protein 3), SQSTM1 (sequestosome 1)/p62 (105), FUNDC1, or OPTN (optineurin).
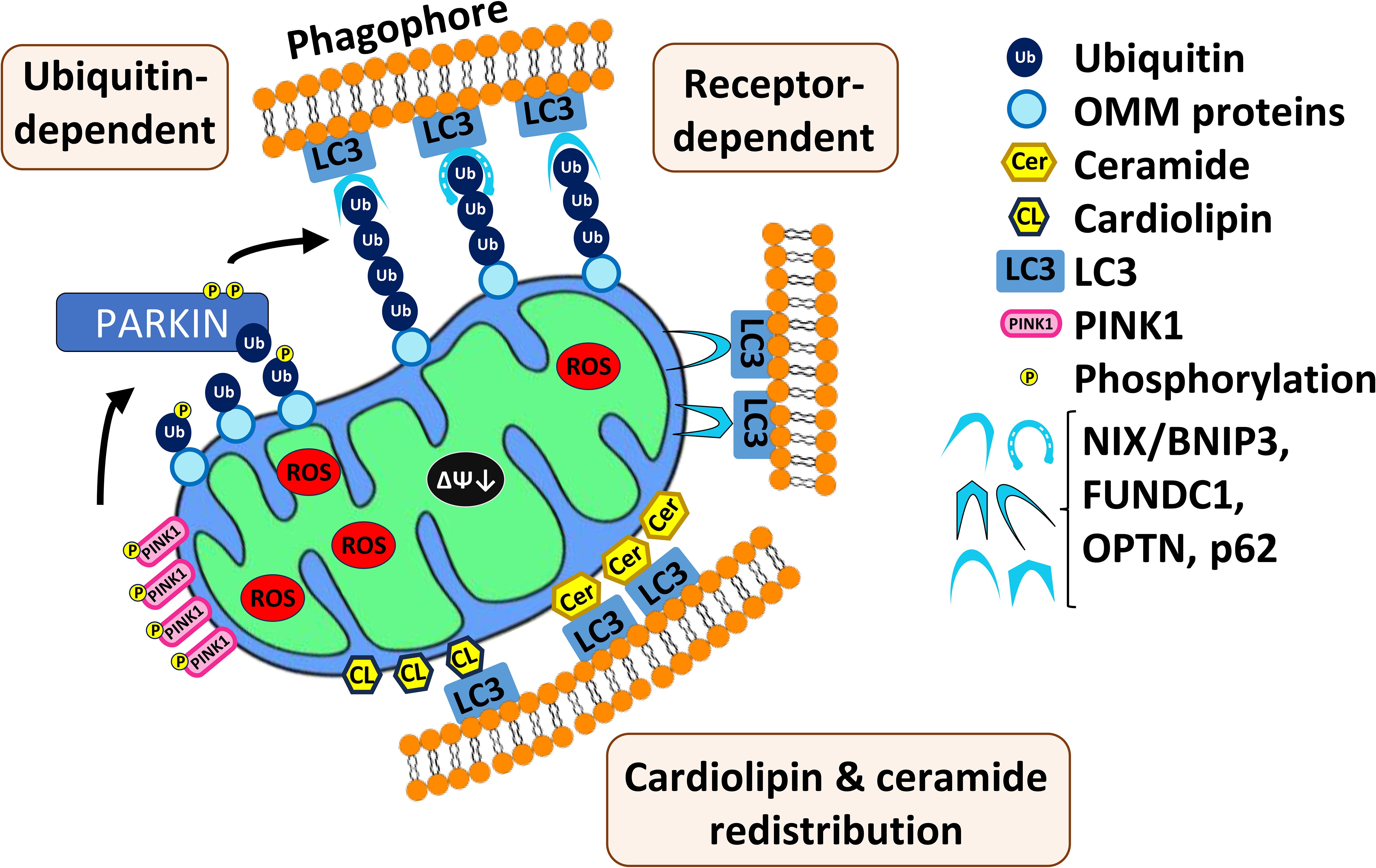
Figure 1. Three main mechanisms of mitophagy in mammals. First, mitochondrial depolarization and impair of protein import cause accumulation of PINK1 on the outer membrane of mitochondria (OMM). PINK1 auto- and transphosphorylates and other targets such as Parkin, which then catalyzes transfer of ubiquitin to various mitochondrial-resident transmembrane proteins. This allows recognition by the autophagosomal sequestration machinery. Second, multiple mitophagic receptors (NIX/BNIPS, FUNDC1, OPTN, p626) are also able to recognize the polyubiquitinated substrates and recruit autophagic machinery. Third, relocalization of cardiolipin and ceramide to the outer membrane of mitochondria where they interact with autophagic machinery.
Mitophagy can also be induced by redistribution of lipid species (Figure 1). For example, cardiolipin is a phospholipid found in the inner mitochondrial membrane (106). During mitophagy, cardiolipin is resorted to the outer mitochondrial membrane and may play a role in recognition of damaged mitochondria and recruitment of the autophagosome (107). Additionally, C18-ceramide, a key component of sphingomyelin, which is a major plasma membrane lipid, has been associated with mitophagy (65). Depletion of C18-ceramide by genetic disruption of the enzyme responsible for its biosynthesis, CerS1, compromised sodium selenite-mediated mitophagy. Additionally, C18-ceramide was shown to relocalize from the endoplasmic reticulum to the outer membrane of mitochondria where, like cardiolipin, it interacts with MAP1-LC3 (65). Cardiolipin and CerS1/C18-ceramide synthesis are associated with tumor suppression, therefore their lack of expression facilitates AML tumorigenesis (65, 108, 109).
Mitophagy can serve as a pro-survival pathway for AML cells by eliminating damaged mitochondria, reducing signaling that may trigger programmed cell death pathways (110, 111). Consistent with this, high expression of mitophagic receptors, including SQSTM1, is associated with increased AML cell survival, contributing to poorer patient outcomes (100). This led to interest in mitophagic receptors as a target for clinical development. SQSTM1 knockout decreased AML proliferation in multiple human and murine AML cell lines. Deletion of this gene also impairs myeloid leukemia progression and prolongs survival in mice (100). Another study similarly found that the downstream SQSTM1 protein (also called p62) and another mitophagic receptor, BNIP3L, increase leukemic cell survival in human AML (112). When bound to the outer membrane of damaged mitochondria, p62 and BNIP3L promote autophagy by recruiting autophagosomes (113). Knockdown of either protein compromised mitochondrial quality and sensitized AML cells to mitochondria-targeting therapeutics. When ROS levels become too high, LSCs induce mitophagy to limit ROS production, or else they will be pushed out of quiescence and cell death pathways will be activated (61, 86). However, high levels of mitophagy in LSCs and bulk AML cells induce multiple cell death pathways (see section 3.4) (65, 114). Therefore, mitophagy plays a dual role in therapeutic approaches: as a resistance mechanism and potential therapeutic target, requiring precise regulation and balance of mitophagy. For example, the pro-autophagy protein, p62, is also a downstream target the of survival and inflammatory pathway, NF-kB (115, 116). Activated p62 upregulates the anti-autophagy and pro-autophagy regulators, mTORC1 and NRF2, respectively. mTORC1 promotes cell growth, inhibits autophagy, and increases metabolism, including glycolysis and lipogenesis, whose upregulation has been implicated in driving carcinogenesis (117, 118). However, activating the antioxidant regulator, NRF2, induces mitophagy (119). This phenomenon highlights the tight regulation of mitophagy that leukemic cells undergo to survive.
In addition to metabolic dysfunction, mitochondria in AML cells also show abnormalities in their shape and structure. Depending on the genetic mutation in the AML cells, many ultrastructural parameters, including mitochondrial volume, the number and diameter of cristae, and the number of matrix granules vary (120). Unsurprisingly, these changes affect the cells’ respiratory profiles including ROS production, basal respiration, proton leak, ATP production, and spare respiratory capacity. Compared to other leukemic cell lines, OCI-AML3 (a representative AML cell line), had more mitochondria and larger cristae. Despite this, OCI-AML3 displayed comparatively lower oxygen consumption rate, indicating inefficient OXPHOS (120). Some studies have argued that the increased mitochondrial mass and number, along with low reserve capacity, a partially depolarized membrane, and the other problems observed, indicate that AML cells’ mitochondria are generally dysfunctional (4, 114). It is also speculated that the larger mitochondrial network observed may be required for other anabolic growth processes (4). Exploiting AML’s dysfunctional mitochondrial characteristics and the tight regulation of mitophagy is explored as a promising avenue for leukemia treatment.
These changes in mitochondrial structure also limit mitophagy, as mitochondria are generally too large to be effectively engulfed for autophagic degradation, so they must be reduced to smaller, more digestible pieces. This is one reason that effective mitophagy requires mitochondrial fission, a process in which proteins in the inner mitochondrial membrane disperse the organelle into smaller pieces. Mitochondrial fission has a counterpart, fusion, wherein smaller mitochondria merge. Fission and fusion are key to the maintenance of mitochondrial quality, as they allow damaged mitochondria to mix their contents to dilute the damage or, by mechanisms that remain unclear, segregate the damaged material into heavily damaged organelles for turnover. The most important proteins for mitochondrial fusion are MFN1 (Mitofusin 1), MFN2 (Mitofusin 2), and OPA1 (optic atrophy 1), while mitochondrial fission is mediated by the master regulator DRP1 (dynamin-related protein 1) and the DRP1-recruiting protein, FIS1 (118). Both DRP1 and FIS1 have been shown to regulate mitophagy as well (86, 121, 122).
3.2 Mitophagic inhibitors as treatment for AMLAs noted above, mitophagy can remove damaged mitochondria, reduce ROS production, improve mitochondrial function, and provide building blocks for continued cell division. As such, it is a natural target for intervention in AML, and several studies have reported positive effects from preventing mitophagy.
The most notable of these involved XRK3F2, an anti-leukemic compound that exerts its effect by impairing the activity of SQSTM1/p62 (123). XRK3F2 treatment causes accumulation of p62 on the mitochondria but reduced colocalization of mitochondria with MAP1-LC3, suggesting that autophagosome recruitment had failed. Consequently, ROS was upregulated, and cell death was triggered. Most importantly, this cytotoxic effect is selective to LSC as neither murine nor human HSCs exhibited significant death after treatment with XRK3F2 (123).
Another promising candidate is the small molecule, S1g-2 (124). This compound was originally discovered in a screen for novel inhibitors of Hsp70, a chaperone that is often overexpressed in drug-resistant CML cell lines (125). S1g-2’s mechanism of action involves binding to Hsp70 (Figure 2), disrupting its interaction with the Bcl-2-like protein Bim, and blocking the oncogenic activation of several proteins, including AKT, Raf-1, and eIF4e. Treatment with S1g-2 induced apoptosis and reduced in vivo burden of CML cells (125). Recently, it has been shown that under stress, the Hsp70-Bim protein complex translocates Parkin and TOMM20 together, leading to TOMM20 ubiquitination and subsequent mitophagic activation (124). Disrupting the interaction between Hsp70 and Bim with S1g-2 blocks mitophagy and indirectly activates apoptosis. As mitophagy is upregulated in both myeloid leukemias, the novel role of Hsp70-Bim in mediating mitophagy may extend S1g-2’s potential to use in AML as well.
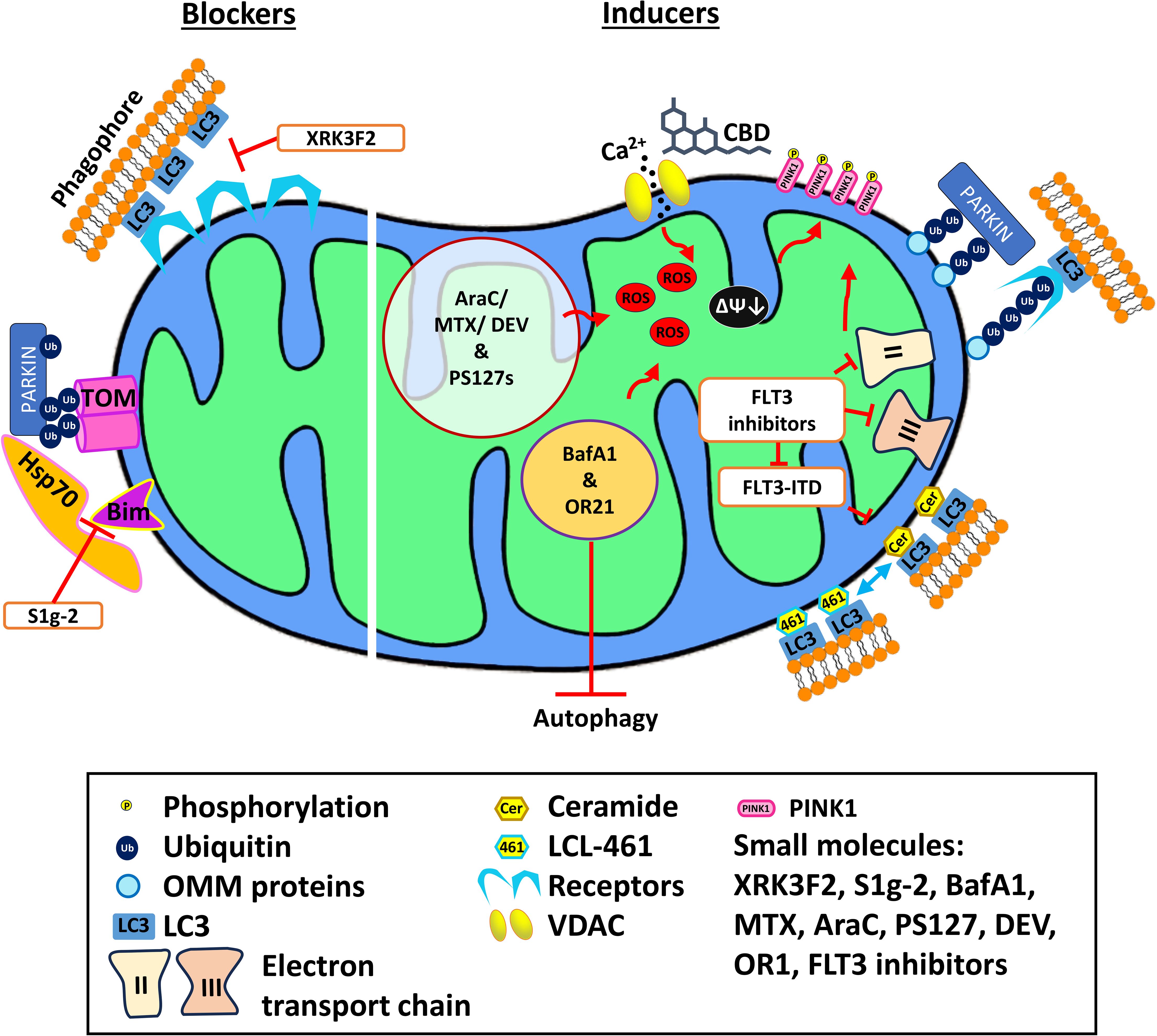
Figure 2. Mechanism of action of various mitophagy blockers and inducers. Mitophagy blockers include XRK3F2 and S1g-2, which binds to the ZZ domain of p62 and Hsp70, respectively, preventing mitophagy. Mitophagy inducers including cannabidiol, PS127 family compounds, BafA1, and OR21 exert their effect by inducing ROS production, thus activating mitophagy. Meanwhile, LCL-461 is a mitochondria-targeted ceramide analog that binds to LC3B and recruits autophagosomes. AraC, cytarabine; BafA1, Bafilomycin A1; DEV, devimistat; MTX, methotrexate; VDAC, voltage-dependent anion channel.
3.3 Mitophagic activators as treatment for AMLWork in our lab to identify compounds that may stimulate mitophagy and improve chemotherapeutic treatment has focused on the PINK1-Parkin axis. We used a high-throughput, phenotypic screening in Caenorhabditis elegans to search for compounds that increased accumulation of a GFP-tagged C. elegans ortholog of PINK1 on mitochondria (126). One advantage of using this system is that it allows rapid removal of compounds that are biochemically effective but have substantial toxicity; dead worms generally do not produce GFP-labeled mitochondria. Using this approach, we identified several compounds that promote PINK-1 accumulation and stimulate mitophagy. The most effective compound and its analogs, called the PS127 family, induced ROS production and impaired mitochondrial respiration only in leukemic cells, and caused accumulation of mitophagic and necroptotic markers. They also activated apoptosis and ferroptosis. Finally, treatment increased survival of mice engrafted with human AML cells (114). The addition of the ROS scavenger, N-acetyl cysteine, significantly rescued PS127 family-induced cytotoxicity, indicating that AML cells are sensitive to increases in ROS. Other labs have also successfully explored whether increasing ROS could be an effective means for inducing leukemic cell death (2, 67, 127).
Increases in ROS can also result from the inhibition of ETC complexes, and by extension, OXPHOS. The ETC Complex I inhibitor, IACS-010759, was shown to increase mitochondrial fission and mitophagy upon treatment in AML cell lines (67). Interestingly, OXPHOS inhibition also promoted the transfer of mitochondria from cocultured mesenchymal stem cells, which rescued cells from apoptosis. To improve treatment, synergy with another ETC Complex I inhibitor, mubritinib, was explored (2). Alterations to other key metabolic pathways, such as fatty acid oxidation, are of interest when exploring AML chemotherapeutics. Similarly to IACS-010759, introduction of the short-chain fatty acid sodium propionate induced ROS, mitochondrial fission, and lethal PINK1- and ATG5-mediated mitophagy via ferroptosis and apoptosis (127). When AML mice were treated with sodium propionate, an anti-leukemic immune response was observed and overall survival increased (127).
Another group of compounds drawing attention for their ability to target mitochondria in AML are cannabinoids, particularly tetrahydrocannabinol (THC) and cannabidiol (CBD), which have been used commonly for palliative care (Figure 2). Each drug and extract has been shown to have antitumorigenic activity (128–130). Both cannabinoids bind to the cannabinoid receptors, CB1 and/or CB2, which are often expressed in AML cells (131). Binding of THC to CB receptors triggers apoptosis in both ALL and AML cell lines, but this response is thought to depend on lymphoid differentiation markers that vary amongst patient cohorts, suggesting that a personalized approach would be needed for this course of treatment to be viable (131). A similar outcome has been observed with CBD, which induces cell death in ALL and CML cell lines (132). Interestingly, CBD treatment appears to have pleiotropic, dose-dependent effects. High concentrations (>30 µM) trigger apoptosis, while lower concentrations (10 µM) activate autophagy. CBD also activates the PINK1-Parkin pathway (133). CBD has also been shown to disrupt the mitochondrial membrane potential, so this result is unsurprising (133). Considering that cyclosporin A reverses CBD-induced mitochondrial depolarization, Parkin recruitment is suggested to result from the formation and opening of the mitochondrial permeability transition pore (133). In CML, where CBD appears to cause similar effects on mitochondria and mitophagic activation, CBD showed synergy with the tyrosine kinase inhibitor, imatinib, the standard treatment for CML, including cytotoxic activity against imatinib-resistant cells (134).
Another mitophagic stimulator showing some promise in pre-clinical testing is the ceramide analog LCL-461 (Figure 2). This drug kills AML patient treatment-resistant blasts with an FLT3-ITD mutation (109). This mutation suppresses ceramide metabolism in AML patients, particularly the production of CerS1-generated C18-ceramides. C18-ceramide allows the execution of lethal mitophagy (109) and is currently thought to be incorporated into the outer mitochondrial membrane by an elegant dance (135). First, cell stress causes dissociation of p17/PERMIT from the mitochondrial fission protein Drp1, allowing the latter to trigger fission and freeing the former to find a new binding partner, in this case CerS1. This relocalizes CerS1 from the endoplasmic reticulum to mitochondria and facilitates C18-ceramide incorporation into the outer mitochondrial membrane by binding to MAP1-LC3 (135). This interaction signals for autophagosomes to execute lethal mitophagy (65, 109).
Treatment effectiveness for multi-kinase inhibitors with effects on FLT3 (e.g., sorafenib or crenolanib) depends on reactivation of C18-ceramide production. Understandably, reducing C18-ceramide synthesis by inhibiting CerS prevented these compounds from inducing AML cell death (109). Sorafenib was later found to inhibit both Complex II and Complex III of the mitochondrial ETC, disrupting mitochondrial membrane potential (136). This dual inhibition stabilizes PINK1 on the outer mitochondrial membrane, leading to mitophagic activation. It is also possible that the mechanism of action for the previously-mentioned C6-ceramide-mediated initiation of mitophagy operates through conversion of C6-ceramide to C18-ceramide (109), a conversion that is known to occur (137). Interestingly, mutation of ASXL1 in AML also causes low-levels of mitochondria-derived vesicles that are implicated in mitochondrial repair and mitophagy (120). This presents the intriguing possibility that LCL-461, or a similar ceramide analog, may also work as a novel anticancer treatment strategy in patients with this type of mutation as well.
Finally, hemin, a physiological erythroid maturation stimulator, was found to induce mitochondrial sequestration in enlarged autophagic vacuoles in the CML cell line K562 (138). Upon treatment with hemin, mitochondria were depolarized, although viability was not compromised. However, localization of the apoptotic protein Nix onto the mitochondrial membrane did trigger mitophagic activation (138). While this was observed in K562, the effect of hemin may translate to other leukemia models for therapeutic purposes and merits further investigation.
3.4 Mitophagic induction improves outcome when combined with standard clinical treatmentIt has often been observed that single-target drugs lead to acquired resistance amongst AML cells (e.g., IDH1 treatments, FLT3 inhibitors, etc.) rendering them less effective or even completely useless. Remission is infrequent and resistance is commonplace. For this reason, AML treatment commonly utilizes drug combinations rather than monotherapies. One burgeoning idea is to combine existing chemotherapeutic treatments with mitophagic regulators. Although inhibiting mitophagy can stimulate cell death in AML, activating this pathway has been subject to more study (114, 139, 140). While it initially seems counterintuitive, excessive activation of mitophagy can reduce survival of leukemia cells (114, 139). Early results suggest that these combinations will be more effective and have reduced off-target effects.
For example, the combination of araC and mitoxantrone (a topoisomerase inhibitor) with devimistat, a CAC cycle inhibitor that induces mitochondrial ROS production, triggers mitophagy (141). Combining devimistat with chloroquine sensitizes AML blasts derived from patient samples. Interestingly, devimistat-induced mitophagy via ROS is independent of PINK1/Parkin (141). Another promising outcome from this study was that increased patient age correlated well with the level of mitochondrial ROS induced by devimistat, suggesting that this treatment may be effective in older patients who often have few treatment options (141). Indeed, mouse AML models with three different baselines of mitochondrial membrane potential showed different responses to devimistat, with a negative correlation between the two variables. Early trial of these drug combinations showed promising results, with a 50% rate of complete remission in patients with relapsed or refractory AML (142). A second study (NCT#02484391) has been conducted, but maintenance devimistat was unfortunately only administered in 2 of 21 responders (141). A third trial (NCT#03504410) complicated this analysis, as patients had poor outcomes and no significant difference between the devimistat treatment and control group were seen, leading to termination of the study (143). Unfortunately, devimistat-treated AML cells respond by increasing their dependency on gluconeogenesis, suggesting a mechanism of metabolic escape in AML that may limit treatment viability (141).
The novel PINK1 Stabilizing compounds discovered in our lab displayed promising synergy when combined with existing chemotherapeutics such as doxorubicin or 6-mercaptopurine in the treatment of AML cells (114). These strong synergistic interactions were highly specific to AML cells, as compared to healthy PBMCs, and effectively killed AML cells that were otherwise resistant to treatment. Interestingly, the synergy and selectivity of PINK1-stabilizing compound in combinations with standard AML treatments were higher than the combination of araC and doxorubicin under similar conditions. Treatment combinations of PS127 compounds with conventional chemotherapeutics were also effective against primary AML cells, although we observed interesting differences in effectiveness that appeared to correlate with AML subtypes (e.g., de novo vs. secondary AML, high or low percentage of blast cells, etc.) (114). While further development of these compounds is necessary, they show strong proof-of-concept for this approach.
3.5 Inhibiting autophagy while simultaneously inducing mitophagy reduced AML survivalIntriguingly, some evidence suggests that inducing lethal mitophagy can be even more effective if autophagy is simultaneously inhibited (84, 85). As has been seen in numerous other conditions, failure of proper autophagosomal formation during mitophagy shifts cells to an apoptotic fate (65, 144). For example, the pharmacological agents bafilomycin A1 (BafA1), chloroquine, and its derivative, Lys05, block autophagy by preventing lysosomal acidification (84, 85). Treatment with any of the three inhibitors increased mitochondrial content under hypoxic conditions (where mitophagy is activated) in AML cells, but not in normal hematopoietic cells. Unlike the other two drugs, BafA1 also exerted its effect in normoxia (84). BafA1 treatment reduced basal and maximal respiration in AML cells, consistent with previous findings that BafA1 induces OXPHOS uncoupling and mitochondrial depolarization (84, 145). ROS production and PINK1 stabilization were also observed in AML cells after treatment with BafA1 (Figure 2). This indicates that BafA1 disrupts mitochondrial function and induces the first steps of mitophagy while preventing completion of mitochondrial degradation. BafA1 treatment was also effective against LSCs. This promising combination also showed greater efficacy in reducing the in vivo tumor burden in mice engrafted with human AML or ALL cell lines (85, 146). Treating araC-resistant AML cells with BafA1 did not restore sensitivity to the chemotherapeutic (147). However, combined treatment of cells with BafA1 and commercial chemotherapeutics (arsenic trioxide (ATO), retinoic acid (ATRA), or araC) induced leukemic cell death, indicating a potential for therapeutic application (148, 149).
Similarly, combination of the hypomethylating agent OR21 with venetoclax significantly increased apoptotic induction through reduction of the mitochondrial homeostatic protein VAMP7, a SNARE family protein responsible for autophagosome formation (150). The combination of venetoclax and OR21 also significantly induced ROS production and mitochondrial damage (Figure 2). A xenografted mouse model treated with this combination showed significantly increased survival compared to single-drug treatment, without weight loss or severe toxicity (150).
4 Omics and functional assays to identify mitochondrial druggable proteinsAs mentioned above, many AML treatments act by disrupting mitochondria to ultimately induce cell death, highlighting the value of probing the structure and function of this organelle for identifying additional chemotherapeutics. Potential drug targets may include mitochondrial translation, ROS production, apoptosis, and energy production (49, 83, 151). However, these are not ‘one size fits all’ therapeutic targets. For example, inducing ROS production can trigger autophagy-induced cell death, but it can also promote oxidative DNA damage favoring carcinogenesis or drug resistance (152–154).
Genetic and transcriptional profiling of AML cells has permitted AML classification based on their cytogenetic mutations and predicting leukemogenesis (155–157). Identifying patterns of co-mutations (whether gain or loss of function) is a useful tool for evaluating key mutations driving relapses (158). Using high-throughput sequencing technology, genetic mutations, gene expression, and epigenetic marking in leukemic cell lines, tumor samples and non-cancerous tissue samples were identified, published, and stored in The Cancer Genome Atlas and LeukemiaDB (159, 160). These databases significantly accelerated the development of diagnostic markers and targeted therapies.
Although transcriptional changes generally match genetic mutations, there are occasional differences, highlighting the importance of evaluating both in the study of cancers (161). Transcriptomics can shed light on gene expression levels and gene functions, which can be used to predict AML progression, relapse risk, and its response to treatment. For example, transcriptional profiling studies have shown that changes in expression of S100A8 and S100A9 alter sensitivity to venetoclax and gilteritinib (162, 163). This profile can also be used to predict overall survival as a function of LYPD3 expression (164). Nonetheless, patients with similar genetic mutations may respond differently to chemotherapy, as the effects of individual mutations are difficult to predict, in part due to other mutations and metabolic variations (165, 166).
The transcriptomes of drug-resistant lines have also been investigated to uncover genes responsible for drug resistance and are subsequently targeted to regain drug sensitivity. This was shown by the identification of mitochondrial translation genes whose disruption could restore sensitivity to venetoclax in otherwise resistant AML cells using a CRISPR screen (163, 167). Additionally, the Cap Analysis Gene Expression (CAGE) transcriptomic approach identified that various genes implemented in cell adhesion and actin polymerization were differentially regulated in OXPHOS-inhibitor resistant patient cells. CAGE also helped identify that mitochondrial fission followed by removal of damaged mitochondria by mitophagy facilitates resistance to OXPHOS inhibitors (67). Importantly, transcriptional profiling is likely to be a key contributor to the development of personalized treatments for AML, a long-sought goal that is beginning to come to fruition and is showing positive results in small clinical trials (168).
Unfortunately, research suggests that transcriptional profiling may also be insufficient for developing personalized medicine and that proteomic analysis could be necessary as well (169). In particular, proteomic profiling of bone marrow cells showed post-translational changes in protein abundance, despite the same levels of RNA transcripts (170). Furthermore, post-translation modifications affect protein functions required for cellular signaling and can further differentiate AML types. For example, changes to phosphorylation profoundly affect AML cell biology, and also need to be taken into account (168, 170–173). Treatment with an FLT3 inhibitor promotes phosphorylation of proteins related to autophagy and mTOR signaling, pinpointing the synergistic cytotoxicity of FLT3 and autophagic inhibitor (173). In attempts to find combinatorial treatments, proteomic and phosphoproteomic (protein phosphorylation profiles) features rationalize cell responses to co-treatments. Proteogenomics identified a specific mitochondrial protein pattern, termed C-Mito, that identifies hypersensitized AML cells to venetoclax and ETC Complex I inhibitor treatment (174). C-Mito provides additional classification of AML based on mitochondrial protein expression not reflected in the classical cellular differentiation markers. Notably, C-Mito-positive patients have a poorer overall probability of survival and worse remission rates than non-C-Mito patients (174).
Although several studies identified potential drug targets using single omics approaches, integrating multi-omics analyses and functional studies may be more promising for enabling a thorough understanding of AML pathogenesis and validate mechanisms of action of drugs. But these data may also enable the identification of actionable targets for treatment. For example, somatic mtDNA analysis of AML cells revealed mutations in genes encoding ETC Complex I, III, or IV (175). As previously discussed, many drugs inhibiting ETC examined in clinical trials, such as IACS-010759 and mubritinib (2, 151).
Transcriptomic, proteomic, and lipidomic approaches also identified a novel druggable target for AML that exploits the regulation of fatty acid oxidation by the mitochondrial deacetylase, Sirtuin-3 (SIRT3). Specifically, SIRT3 inhibition suppresses fatty acid oxidation, causing reduction of OXPHOS and ATP level, which leads to LSCs vulnerability to lipotoxicity (176). LSCs upregulate cholesterol metabolism to tolerate lipid accumulation causing cell death and lipotoxicity, suggesting potential mechanism to sensitize LSCs to SIRT3 inhibition.
5 ConclusionThe metabolic reprogramming required to support AML, favoring OXPHOS and fatty acid oxidation, precariously balances their mitochondrial activity, sensitizing them to any further mitochondrial disruption. This highlights the value of targeting mitochondria for therapeutic intervention. Although the dual role of mitochondrial metabolic pathways may currently pose a challenge for the effectiveness of current commercial treatments, they are the most likely route for moving forward in developing effective, long-term treatments for AML.
Targeting mitophagy has shown desirable effects in providing a new treatment modality and in abrogating resistance to conventional chemotherapeutics in AML cells. Tests of both mitophagic blockers (XRK3F2, S1g-2) and inducers (PS compounds, Hemin, CBD, LCL-461, devimistat) have shown positive effects in specifically targeting leukemic cells with minimal effects on healthy hematopoietic cells. These results, while initially surprising, suggest that either method can lead to the same end: damaged and dysfunctional mitochondria and apoptotic initiation. Clearly further investigation is needed to determine which course of treatment is most effective, but we anticipate that this research will demonstrate that optimized treatments will vary based on each patient’s genetic and metabolic state. For example, inducing mitophagy in AML has been most effective at destroying cancer cells that harbored FLT3-ITD mutations. It should be noted, however, that most of the studies testing this approach have used cells harboring this mutation. It is imperative that a wider panel of AML mutations be tested with combinations of mitophagic activators or inhibitors alone and with conventional chemotherapeutics, to evaluate the practicality of this therapeutic approach more generally.
Author contributionsET: Investigation, Visualization, Writing – original draft, Writing – review & editing. MD: Investigation, Writing – review & editing. BM: Investigation, Writing – review & editing. AR: Investigation, Visualization, Writing – review & editing. NK: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIH NCI R21CA280500 and NIGMS R35GM129294 to NVK and T32 GM139801 to MD.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statementThe author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Blum W, Matthews V. Fast Facts: Acute myeloid leukemia. Abingdon, Oxford OX14 3LN, UK: Karger (2018).
2. Baccelli I, Gareau Y, Lehnertz B, Gingras S, Spinella J-F, Corneau S, et al. Mubritinib targets the electron transport chain complex I and reveals the landscape of OXPHOS dependency in acute myeloid leukemia. Cancer Cell. (2019) 36:84–99.e8. doi: 10.1016/j.ccell.2019.06.003
PubMed Abstract | Crossref Full Text | Google Scholar
3. Panina SB, Baran N, Brasil Da Costa FH, Konopleva M, Kirienko NV. A mechanism for increased sensitivity of acute myeloid leukemia to mitotoxic drugs. Cell Death Dis. (2019) 10:617. doi: 10.1038/s41419-019-1851-3
PubMed Abstract | Crossref Full Text | Google Scholar
4. Nelson MA, McLaughlin KL, Hagen JT, Coalson HS, Schmidt C, Kassai M, et al. Intrinsic OXPHOS limitations underlie cellular bioenergetics in leukemia. eLife. (2021)
留言 (0)