Renal function relies on the interplay between glomerular filtration and tubular processes. The glomerular filtration rate (GFR), the sum of all single-nephron GFRs (SNGFRs), is a key indicator of kidney function. Glomerular filtration adapts dynamically to physiological needs, such as transient hyperfiltration after high-protein meals [1, 2].
Two types of hyperfiltration are recognized: absolute and relative.
Absolute hyperfiltration occurs when SNGFR increases in kidneys with a normal number of nephrons. This can be observed in physiological states, such as pregnancy, or pathological conditions.
Relative hyperfiltration occurs when SNGFR increases in the remaining nephrons following nephron loss, such as in congenital solitary kidneys, prematurity, intrauterine growth restriction (IUGR), or nephron reduction due to surgery or disease. Although initially adaptive, this response eventually becomes pathological in chronic kidney disease (CKD), contributing to disease progression. Pathological hyperfiltration is also observed in conditions such as diabetes, obesity, malignancy, sleep apnea, and high-altitude exposure [1, 3].
The mechanisms underlying hyperfiltration are well-characterized in conditions like diabetes and obesity. Key factors include increased glomerular capillary pressure, glomerular enlargement, and disrupted tubuloglomerular feedback. Vascular regulation involves angiotensin II-induced efferent arteriolar constriction, with agents like angiotensin-(1–7), nitric oxide, and COX-2-derived prostanoids causing afferent arteriolar dilation. Endothelin-1 affects both arterioles, and its systemic effects can be mitigated by endothelin receptor A blockade [4] (J Am Soc Nephrol. 2000;11:1498–504).
In diabetes, excessive glucose filtration enhances SGLT2 activity, leading to increased proximal tubular sodium reabsorption, reduced solute delivery to the macula densa, and afferent arteriolar vasodilation [5]. Similarly, in obesity, renal compression by adipose tissue activates the renin-angiotensin system (RAS) and the sympathetic nervous system, exacerbating sodium retention [6].
Clinically, hyperfiltration induces mechanical stress on the glomeruli, leading to basement membrane expansion, podocyte injury, increased albuminuria, and progression to glomerulosclerosis and tubular damage [7, 8]. In early diabetic nephropathy, hyperfiltration is associated with an increased tubular metabolic load and oxygen consumption.
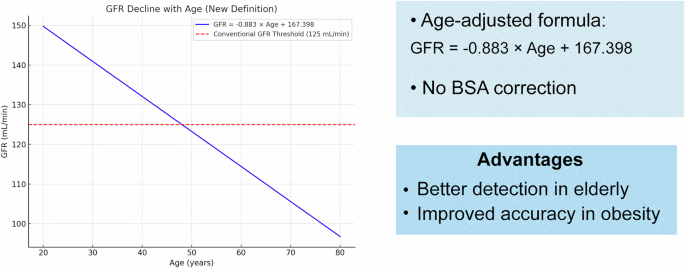
Despite its importance, a universally accepted definition of hyperfiltration is lacking. Commonly reported thresholds range from 130 to 140 mL/min/1.73 m² in individuals with two functional kidneys, corresponding to renal function exceeding two standard deviations above the mean GFR in healthy individuals. However, fixed thresholds fail to account for factors such as sex, ethnicity, nephron number at birth, or age-related GFR decline [9].
Therapeutically, the initial GFR reduction observed with renoprotective drugs reflects hyperfiltration correction. Agents like SGLT2 inhibitors and incretin-based therapies may benefit high-risk patients, even in the absence of CKD [10]. Early detection and intervention in hyperfiltration-associated conditions are essential to prevent or delay kidney disease progression.

留言 (0)