
記住我
• The lactate metabolism and transport carried out in neurons and glial cells may influence the development of epilepsy by regulating protein lactylation levels.
• Lactate may mediate epilepsy-related neuronal loss by promoting HMGB1 lactylation.
• Ischemic stroke may promote HMGB1 lactylation by activating HIF-1, which can promote a shift in the mode of cellular energy acquisition from oxidative phosphorylation to glycolysis, thereby inducing post-stroke epilepsy.
• Histone deacetylases may affect protein lactylation by regulating the transcriptional activity of HIF-1.
• Histone deacetylase inhibitors may combat post-stroke epilepsy by modulating hypoxia-induced protein lactylation.
1 IntroductionEpilepsy, as a common chronic disease of the nervous system, has affected more than 70 million people of all ages worldwide, with the number continuing to rise annually (Eugen et al., 2019). The factors inducing seizures are diverse and multifaceted. Ischemic stroke is one of the common causes of epilepsy. Post-ischemic stroke epilepsy accounts for approximately 9% of all epilepsy cases, a figure slightly lower than that caused by cerebral hemorrhage. However, the growing number of individuals affected by ischemic stroke each year highlights its significant impact on public health (Lanqing et al., 2021; Carolina et al., 2021). The hypoxic environment caused by ischemic stroke often forces brain tissue to obtain energy through glycolysis, thereby producing a large amount of lactate.
Lactate in the brain may affect the progression of various neuropsychiatric diseases through its roles in learning, memory, and emotional regulation. This process may be related to lactate-mediated post-translational modifications of proteins (Hagihara et al., 2021). Lactylation (La), a novel type of post-translational protein modification, was identified by Zhang et al. (2019). With the ongoing research into protein lactylation modification in recent years, HAGIHARA H et al. found that lysine lactylation (Kla) may be prevalent in neurons and glial cells in the brain.
Furthermore, the level of lysine lactylation is regulated by lactic acid levels, neural excitation, and social defeat stress (Hagihara et al., 2021). It suggests that increased neuronal excitability may be closely related to the level of protein lactylation in neurons.
Abnormal excitation of neurons is a key driver of epileptogenesis. Ischemia/reperfusion injury (I/R) can induce epileptogenesis by mediating abnormal excitation of neurons (Paudel et al., 2020; Esih et al., 2021; Endres et al., 2022). It can be speculated that ischemia-induced stroke may lead to increased glucose metabolism in the brain, resulting in the production of large quantities of lactate, which can be transformed into acetyl groups to participate in the acetylation of proteins related to neuronal excitation and thus cause post-stroke epilepsy (Hagihara et al., 2021; He et al., 2023). That is, protein lactylation may be a potential regulatory target for post-ischemic stroke epilepsy. While anti-epileptic drugs can help most patients control their seizures, approximately 30% of individuals with epilepsy do not respond to current clinical medications (Sun et al., 2021). This review mainly focuses on the impact of lactate metabolism and protein acetylation on epilepsy and changes in acetylation modifications controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Furthermore, based on the mechanisms of action of HDAC inhibitors (HDACIs) in regulating acetylation modifications, we discuss the possibility that they could provide a new strategy for treating post-stroke epilepsy.
2 The lactate mediates epilepsy by regulating the functions of neurons and glial cellsSeizures are usually localized in the hippocampus, with temporal lobe epilepsy (TLE) being the most common (Sun et al., 2021). Previous studies have suggested that neuronal hyperexcitability in epilepsy is primarily mediated by neuronal death and neuroinflammation, which is mediated by astrogliosis and microglial activation, and the relationship between neuronal death and neuroinflammation may be complex (Haenisch et al., 2015; Roh et al., 2023). Seizures are usually accompanied by significant glial cell proliferation and loss of neurons, and glial cell proliferation-mediated neuroinflammation often leads to neuronal death (Feng et al., 2019). Protein lactylation modifications have been found in the hippocampus and prefrontal cortex and are strongly associated with neuronal excitability (Hagihara et al., 2021).
Currently, there is insufficient evidence to confirm that protein lactylation directly induces epilepsy by regulating neuronal excitability. However, lactate, as the donor of lactide, can serve as an energy source for metabolic activities in neurons and glial cells during ischemia and hypoxia, potentially influencing the progression of post-stroke epilepsy.
2.1 Glial cells mediate epilepsy by regulating the release of neurotransmittersStudies on status epilepticus suggest that astrocyte and microglial activity changes, which influence critical homeostatic processes—such as synaptogenesis, extracellular ion concentrations, and excitation-inhibition balance—may occur early in epilepsy development. Inhibiting glial cell activity has been shown to reduce susceptibility to epilepsy (Shen et al., 2023). Neuroglia, such as astrocytes, microglia, and oligodendrocytes, may influence neuronal excitability by regulating the production of glutamate, adenosine triphosphate (ATP), and γ-aminobutyric acid (GABA) in the central nervous system, thereby modulating the progression of epilepsy (Shen et al., 2023).
Glutamate is the primary excitatory neurotransmitter in the central nervous system and can induce seizures by increasing synaptic transmission in the hippocampal region. Although the death of neurons caused by cerebral infarction can reduce the synthesis of the neurotransmitter glutamate, previous studies have confirmed that the level of glutamate in the blood and cerebrospinal fluid of ischemic stroke patients is related to the severity of infarction and neurological dysfunction (Nicolo et al., 2019). Research has shown that stroke can mediate neuroinflammatory damage by activating astrocytes’ TGF-β pathway, and the TGF-β signaling in astrocytes may have a pro-epileptic effect (Korotkov et al., 2020). Therefore, astrocytes may induce post-stroke epilepsy by mediating stroke-related glutamate excitotoxicity. Astrocytes primarily take up excessive glutamate from the synaptic cleft through two types of glutamate transporters, such as glutamate–aspartate transporter (GLAST) and glutamate transporter-1 (GLT1). The glutamate, which is transported into the cell by GLAST or GLT1, can be converted into glutamine by glutamine synthetase, which is a major precursor for the biosynthesis of the inhibitory neurotransmitter GABA (Sun et al., 2021; Peterson et al., 2021). Glutamate released by neurons is taken up by astrocytes and converted to glutamine to alleviate the excitotoxicity of glutamate on neurons. Glutamine serves as a source of energy for both glial cells and neurons (Nagy et al., 2018). Recent studies have shown that reducing the expression of nascent proteins and the activity of inward rectifying K+ channel subtype 4.1 (Kir4.1), respectively, leads to a decrease in the expression of GLAST and GLT1 in hippocampal astrocytes, which results in increased extracellular glutamate levels and decreased GABA release, leading to an increased risk of epileptogenesis (Sun et al., 2021; Centonze et al., 2023). Since knocking down nascent protein does not affect the expression of GABA transporter proteins in astrocytes, the reduction in GABA release may be attributed to reduced GABA synthesis (Sun et al., 2021). It has been shown that inhibiting the conversion of putrescine to spermine in astrocytes would lead to more remaining putrescine being available for the synthesis of GABA, thereby effectively reducing epileptic activity (Kovács et al., 2021).
Previous research suggests that compared to GLAST, GLT1 is the main transporter for the uptake of glutamate by astrocytes (Peterson et al., 2021). In the intrahippocampal kainic acid model of TLE, GLT1 regulates the frequency of seizures and the total time spent in seizures by mediating the uptake of most glutamate in the dorsal forebrain (Peterson et al., 2021). GLT1 mitigates ischemic stroke-induced neuroexcitotoxicity by increasing glutamate reuptake in astrocytes, thereby reducing neuronal cell death, which is an important cause of post-stroke epilepsy (Wang et al., 2022). Post-translational modifications of GLT-1, including palmitoylation, ubiquitination, nitrosylation, and succinylation, can regulate the distribution of GLT-1 and the rate at which it transports glutamate (Peterson et al., 2021). Although it has not yet been confirmed that palmitoylation can alter the activity of GLT1, based on reports linking protein palmitoylation to neuronal excitability, GLT1 lactylation may affect the excitability of neurons by regulating the concentration of glutamate between synapses (Hagihara et al., 2021).
Previous studies have shown that calcium ions mediate the release of glutamate and ATP from neurotransmitter-activated astrocytes to promote neuronal firing patterns (Shen et al., 2023; Chen et al., 2023). However, other research suggests that the majority of the neurotransmitter glutamate in the brain is synthesized and released by neuronal cells, with astrocytes playing a role in regulating the concentration of glutamate between synapses through reuptake. Until the concept of “glutamatergic astrocytes” was proposed by DeCeglia et al., the ability of astrocytes to transmit information like neurons was unknown. Glutamatergic astrocytes are located mainly in the hippocampus and express the vesicular glutamate transporter protein 1 (vGLUT1), which specifically loads glutamate into synaptic vesicles and promotes its release into the synaptic gap (de Ceglia et al., 2023). Although vGLUT1-mediated glutamate release may promote the occurrence of epilepsy, it does not prove that the glutamate transported by vGLUT1 is synthesized by astrocytes rather than acquired by astrocytes from the synaptic cleft. Glutamate can not only directly induce epilepsy by mediating the excitotoxicity of neurons but also induce epilepsy related to brain tissue damage by altering the function of inhibitory interneurons. Previous studies have confirmed that brain tissue damage can lead to an increase in synaptic inputs generated by pyramidal neurons, thereby driving an increase in the excitability of surviving hilar interneurons, which in turn leads to the occurrence of post-traumatic epilepsy (Butler et al., 2017). This may be closely related to the glutamatergic synapses formed between pyramidal cells and interneurons in the hippocampus, as a previous study has shown that pyramidal cells may regulate the transition between inhibitory interneurons and disinhibitory interneurons through the glutamatergic synapses formed with interneurons (Tzilivaki et al., 2023).
During status epilepticus, neurons, astrocytes, and microglia release ATP, activating the purinergic receptor P2Y1 on astrocytes and further exacerbating neuronal excitotoxicity. Meanwhile, ATP released from damaged neurons can activate NLRP3 inflammatory vesicles by binding to the ATP-gated ion channel P2X7 in microglia, mediating inflammatory injury in brain tissue (Liu et al., 2022). Glial cells and neurons can sustainably increase neuronal excitotoxicity by releasing excitatory neurotransmitters. However, adenosine kinase synthesized by astrocytes converts ATP to adenosine, alleviating neuronal excitotoxicity by activating presynaptic A1 adenosine receptors, thereby blocking this self-sustaining cycle of neuronal activity (Shen et al., 2023; Boison and Steinhäuser, 2018). Meanwhile, as brain-resident immune cells, microglia catalyze the conversion of ATP to adenosine by expressing CD39, an extracellular ATP/ADP hydrolase encoded by Entpd1, which can reduce seizures (Hu et al., 2023). Although activated microglia may promote the occurrence of epilepsy by releasing glutamate and inflammatory cytokines (such as interleukin-1β, interleukin-6, and tumor necrosis factor-α), inhibiting glutamate uptake, and reducing GABAergic transmission (Shen et al., 2023), the above-mentioned astrocytes and microglia can play a dual role in promoting and inhibiting epilepsy, which may depend on different stages of epileptogenesis.
Previous studies have shown that oligodendrocytes, like astrocytes and microglia, regulate the release and reuptake of neurotransmitters between neuronal synapses, thereby mediating epileptogenesis and development. Oligodendrocytes have been shown to be involved in the regulation of glutamate levels in the brain (Shen et al., 2023).
2.2 Lactate interference with glial cell functionGlucose is the main energy source for the brain, and interfering with the glucose metabolic process may affect brain function. Lactate produced by glycolysis may mediate glial cell activation and neuronal functional impairment by promoting histone acetylation (Pan et al., 2022; Wei et al., 2023). The increase in histone H3K18 acetylation levels in the hippocampus can directly activate the nuclear factor kappa-B (NF-κB) signaling pathway by stimulating the promoters of Rat transcription factor (Rela) and NF-κB, which can enhance the production and release of pro-inflammatory factors and exacerbate the inflammatory damage associated with epilepsy (Wei et al., 2023; Cai and Lin, 2022). This pathological change may be amplified by a positive feedback loop between glycolysis/H4K12la/ pyruvate kinase isozyme type M2 (PKM2). As a key enzyme in the glycolytic pathway, PKM2 can ensure that the cell’s energy metabolism switches from oxidative phosphorylation to glycolysis. The H3K18la mainly mediates the pro-inflammatory activation of microglia through the NF-κB signaling pathway. Microglia activation can exacerbate neuronal functional damage by promoting the formation of NLRP3 inflammasomes and Apoptosis-Associated Speck-Like Protein Containing a Caspase Recruitment Domain (ASC) specks (Pan et al., 2022).
Microglia cells have significantly higher levels of H4K12la at the promoters of hypoxia-inducible factor-1α (HIF-1α), PKM2, and lactate dehydrogenase (LDH), which leads to increased expression of these glycolysis-related proteins and further upregulates H3K18 and H4K12 acetylation (Pan et al., 2022; Wei et al., 2023). While there was no significant difference in H4K12la between the transgenic AD model mice and wild-type mice in the affected areas of their brains, the research suggests that the metabolism of astrocytes and neurons, like microglia, is regulated by lactate (Pan et al., 2022; Barros et al., 2023). Previous studies have suggested that cognitive impairment may be linked to the expression levels and activities of key enzymes in glycolysis, such as LDH, as well as glucose transporter protein 1 (GLUT1) and GLUT3, which are the primary cellular channels for glucose uptake in neurons and astrocytes, respectively (Yang et al., 2024; Wu et al., 2023). It is, therefore, possible that the glucose uptake of astrocytes and neurons may affect brain function by regulating lactate production. Based on the evidence of crosstalk between microglia, astrocytes, and neurons, the functions of starry glial and neuronal cells may also be influenced by protein acetylation, as in microglia (Mayorga-Weber et al., 2022; Mason, 2017). The lactate/H4K12la/PKM2 feedback loop in microglia might continuously promote the acetylation of histones in neurons and astrocytes by providing lactate. Furthermore, as GLUT1 on astrocytes transports more glucose into the cell, aerobic glycolysis gradually replaces mitochondrial oxidative phosphorylation as these cells’ main glucose metabolic pathway. The glucose taken up by neurons via GLUT3 not only participates in oxidative energy supply but also is metabolized by the pentose phosphate pathway to eventually produce glutathione to maintain redox balance (Mayorga-Weber et al., 2022) (Figure 1).
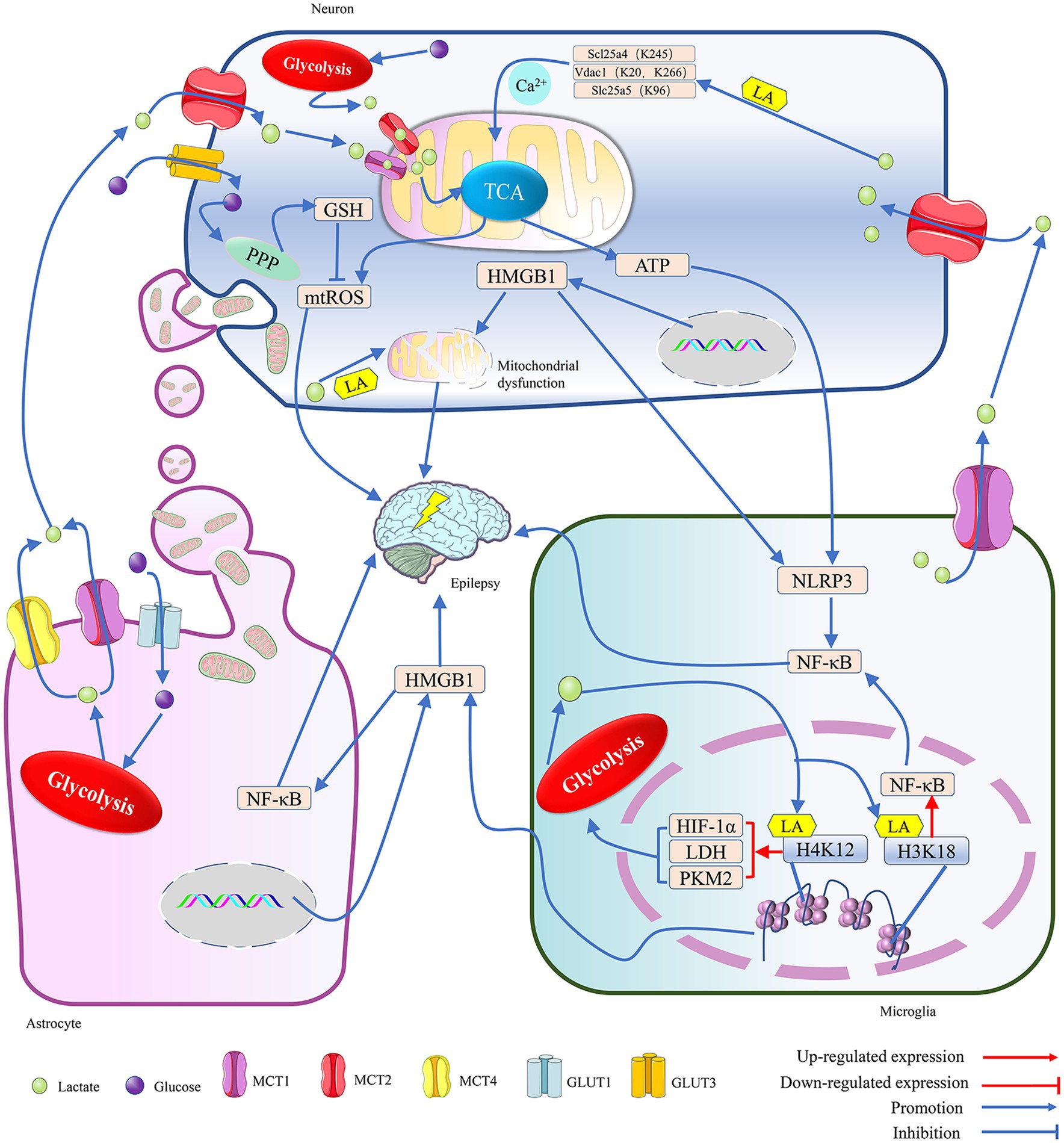
Figure 1. The metabolic crosstalk between neurons and glia involving lactate is implicated in epilepsy. 1. Both glucose and lactate can serve as energy substrates for neurons. Glucose is primarily taken up by neurons via GLUT3, while lactate, mainly provided by astrocytes and microglia, is mostly taken up by neurons via MCT2 during situations of intense stimulation of neural function. In a hypoxic environment, within neurons, the lactate generated from glucose through glycolysis and the lactate transferred into the cell by MCT2 are spread on the mitochondrial membrane by MCT1 and MCT2. There, they penetrate the TCA cycle, and the TCA contributes to the production of a significant amount of ATP and ROS through the promotion of oxidative phosphorylation. The ATP not only supports neuronal excitation but also activates the NLRP3/NF-κB pathway in microglia, leading to epilepsy. The mtROS may cause epilepsy by inducing oxidative stress in neurons, but PPP can reduce it by synthesizing glutathione, an antioxidant. Additionally, lactate can lead to epilepsy by enhancing the lactylation levels of proteins related to mitochondrial function, such as inducing Ca2+ overload-mediated mitochondrial apoptosis by promoting lactylation of key proteins of the Ca2+ signaling pathway, Scl25a4 (K245), Slc25a5 (K96) and Vdac1 (K20, K266). The transfer of HMGB1 from the nucleus to the cytoplasm can help coordinate homophilin lactylation, leading to epileptogenesis by mediating neuronal mitochondrial dysfunction. However, astrocytes maintain normal neuronal physiological activity and reduce epileptogenesis by providing healthy mitochondria to neurons. 2. In astrocytes, glucose taken up by the GLUT1 channel is converted into lactate, which is primarily exported by the MCT1/4 and used as an energy source by neurons. Both microglia and astrocytes can secrete HMGB1, which is a damage-associated molecular pattern and continues to induce the activation of glial cells and the inflammatory damage of neurons, leading to the occurrence of epilepsy. Meanwhile, HMGB1 secreted by microglia can mediate epileptogenesis through activation of the NF-κB pathway in astrocytes. 3. In a hypoxic environment, the lactate generated by glycolysis in microglia not only supports neuronal energy metabolism but also facilitates the lactylation of histones H3K18 and H4K12, which enhances the expression of NF-κB and glycolysis-related genes. NF-κB can trigger the onset of epilepsy, and the enzymes, such as LDH, HIF-1α, and PKM2, can further promote the production of lactate, which then helps in the lactylation of histones, ultimately intensifying the symptoms of epilepsy. Adenosine triphosphate (ATP); glucose transporter 1/3 (GLUT1/3); glutathione (GSH); high-mobility group box 1 (HMGB1); hypoxia-inducible factor 1 alpha (HIF-1α); K (Lysine); monocarboxylate transporter (MCT); mitochondria reactive oxygen species (mtROS); NLR family pyrin domain containing 3 (NLRP3); nuclear factor kappa-B (NF-κB); pentose phosphate pathway (PPP); pyruvate kinase isozymes M2 (PKM2); and tricarboxylic acid cycle (TCA).
Cerebral ischemia can induce the proliferation of astrocytes and stimulate their glycolytic metabolism (Lv et al., 2015), leading to the release of lactate, which can then enter neuron cells through monocarboxylic acid transporter (MCT) to cause an increase in neuronal activity (Barros et al., 2023; Yang et al., 2024; Wu et al., 2023). With the release of lactate from astrocytes, the negative feedback on glycolysis gradually weakens, allowing astrocytes to continuously produce lactate for energy transport to the neuron cells (Barros et al., 2023). Additionally, it has been demonstrated that neurons can uptake lactate released by microglia via MCT2 (Mayorga-Weber et al., 2022). Consequently, microglia and astrocytes may maintain neuron energy metabolism by continuously supplying lactate. This lactate may affect the physiological functions of neurons by promoting histone lactylation in hippocampal tissue (Nagy et al., 2018; Wei et al., 2023) (Figure 1).
MCT1-4 show cell-type-specific distribution. MCT1/4 is primarily located on the membranes of astrocytes, with MCT4 being unique to astrocytes. MCT2 is expressed exclusively by neurons, and microglia mainly rely on MCT1 for lactate transport (Yang et al., 2024; Jády et al., 2016). Lactate/pyruvate is transported between cells via MCTs. The imported pyruvate may enter the mitochondria and preferentially serve as a substrate for the pyruvate dehydrogenase system (PDHc) to provide energy for the cells, while the imported lactate is first oxidized by LDH to produce NADH, which then follows a specific shuttle mechanism to enter the mitochondria. When the shuttling of NADH is impeded, it may prevent the oxidation of lactate (Nagy et al., 2018) as a product of glycolysis; lactate/pyruvate can be used by the mitochondria for oxidative phosphorylation. Astrocytes can release lactate through MCT1/4, which may then enter neuronal cells via MCT2. Finally, the lactate enters the mitochondria through the MCT1/2 channels in the neuron’s cytoplasm (Yang et al., 2024; Wu et al., 2023; Yang et al., 2022), promoting mitochondrial energy metabolism and reactive oxygen species (ROS) production. The mitochondria ROS (mtROS) can induce dysfunction of neuronal mitochondria and damage the structure and function of synaptic elements (Jia et al., 2021). Moreover, the uptake of dysfunctional mitochondria released from glial cells by neurons can also lead to abnormalities in neural discharge (Zhang et al., 2023). Thus, lactate metabolism and mitochondrial function in glial cells can affect neuronal discharge frequency. It has been shown that an increase in the number of mitochondria and a disturbance in their dynamics in the hippocampus can lead to excessive excitation of hippocampal neurons and prolonged epileptic duration in mice (Bebensee et al., 2017; Lee et al., 2022) (Figure 1).
Activated microglia can cause neuronal damage by releasing dysfunctional mitochondria, whereas astrocytes can support neuronal survival by transferring functional mitochondria to them (Jia et al., 2023). Astrocytes might regulate neuronal activity by providing “powerhouses” (mitochondria) and the energy substrate lactate, thereby affecting anxiety-like behavior and cognitive deficits caused by seizures (Wu et al., 2023; Jia et al., 2023). Neurons can take up glucose through GLUT3 and break it down via glycolysis to generate energy, but their reliance on astrocytes for energy metabolism may help them avoid apoptosis induced by high glycolytic rates (Wu et al., 2023). Thus, the regulation of neuronal activity by astrocytes is closely related to mitochondrial function and lactate metabolism. Mitochondria have been shown to transfer between neurons and glial cells, and exogenous mitochondria can cross the blood–brain barrier to reduce ROS release induced by epilepsy, thereby reducing hippocampal neuron loss and glial activation (Jia et al., 2023). Numerous studies have confirmed that mitochondrial dysfunction can lead to increased lactate production through enhanced anaerobic glycolysis (Barros et al., 2023; Yang et al., 2024; Wu et al., 2023), and the lactate may mediate the occurrence and development of epilepsy by promoting protein lactylation modifications (Figure 1).
2.3 The impact of lactate metabolism on neuronal cell deathPrevious studies have suggested that neuronal loss associated with programmed death plays an important role in the development of epilepsy (Liang et al., 2023).
2.3.1 NETosisNeuroinflammation is one of the main causes of neuron loss (Li et al., 2021). Neuroinflammation is characterized not only by the proliferation of glial cells but also by disruption of the blood–brain barrier and migration of peripheral immune cells into the brain. Neutrophils infiltrate damaged brain tissue immediately after ischemic injury, and they can sustain neuronal damage around the infarct by releasing neutrophil extracellular trapping networks (NETs) (Li et al., 2021; Bernis et al., 2023; Byun et al., 2023). NETosis is a form of suicidal death for neutrophils, characterized by the formation and release of NETs, which primarily consist of double-stranded DNA, histones, and granule proteins (Li et al., 2021). During chronic inflammatory damage in the central nervous system, inhibiting the activity of key enzymes in the glycolytic pathway can block the activation of NETosis by reducing lactate levels (Awasthi et al., 2019; Ye et al., 2022; Wang et al., 2018). NETosis may exacerbate neuroinflammation by releasing inflammatory factors to induce neuronal loss in brain tissues after ischemic infarction. The hypoxic environment created by ischemic stroke can induce the occurrence of glycolysis, leading to the production of large amounts of lactate. The lactate may trigger NETosis and contribute to the development of post-stroke epilepsy by enhancing the lactylation of histones in neutrophils that infiltrate the ischemic focus.
2.3.2 CuproptosisRecent research has indicated that neuronal cuproptosis may be an important factor in the initiation and progression of TLE (Yang et al., 2023). Cuproptosis is a mitochondrial proteotoxic stress-dependent mode of regulated cell death (RCD), where the accumulation of copper in cells is crucial (Chen et al., 2023). Ferredoxin 1 (FDX1), a key protein in cuproptosis, can trigger mitochondrial protein lipidization, leading to mitochondrial protein toxicity stress. This form of stress is distinct from copper-induced mitochondrial oxidative stress and the iron-induced oxidative cell death associated with ferroptosis. As mitochondria are the primary targets in cuproptosis, the process includes the disintegration of the mitochondrial membrane and the functional loss of enzymes critical to the tricarboxylic acid cycle (Liu et al., 2022). When oxidative phosphorylation is inhibited, glycolysis is activated as the main energy source for cells (Barros et al., 2023). It seems that the occurrence of cuproptosis may help enhance the level of lactate. In turn, when aerobic glycolysis replaces the aerobic oxidation of sugars as the main energy source for cells, cuproptosis is likely to be suppressed (Xiong et al., 2023). It has been demonstrated that overexpression of FDX1 induces cuproptosis in tumor cells by catalyzing the lipoylation of PDH and α-ketoglutarate dehydrogenase (Schulz et al., 2023). However, HIF-1α can block FDX1-induced cuproptosis by indirectly inhibiting PDH (Chen et al., 2023). PDH promotes the entry of pyruvate into mitochondria and initiates the tricarboxylic acid cycle. Inhibition of PDH activity leads to mitochondrial energy depletion, which activates AMPK to promote cuproptosis (Xue et al., 2023). Copper overload may induce cuproptosis by mediating mitochondrial respiratory chain damage by over-activating the energy sensor AMPK. Cuproptosis can promote the release of the pro-inflammatory mediator High-Mobility Group Box 1 (HMGB1) (Liu et al., 2022; Xue et al., 2023) (Figure 2). It appears that cuproptosis-induced mitochondrial dysfunction may inhibit the exacerbation of cuproptosis by promoting the production of lactate, which has been shown to activate HIF-1α. HIF-1α may antagonize the effects of FDX1-mediated lipid acylation of PDH by inhibiting the activity of PDH to inhibit cuproptosis (Jiang et al., 2021).
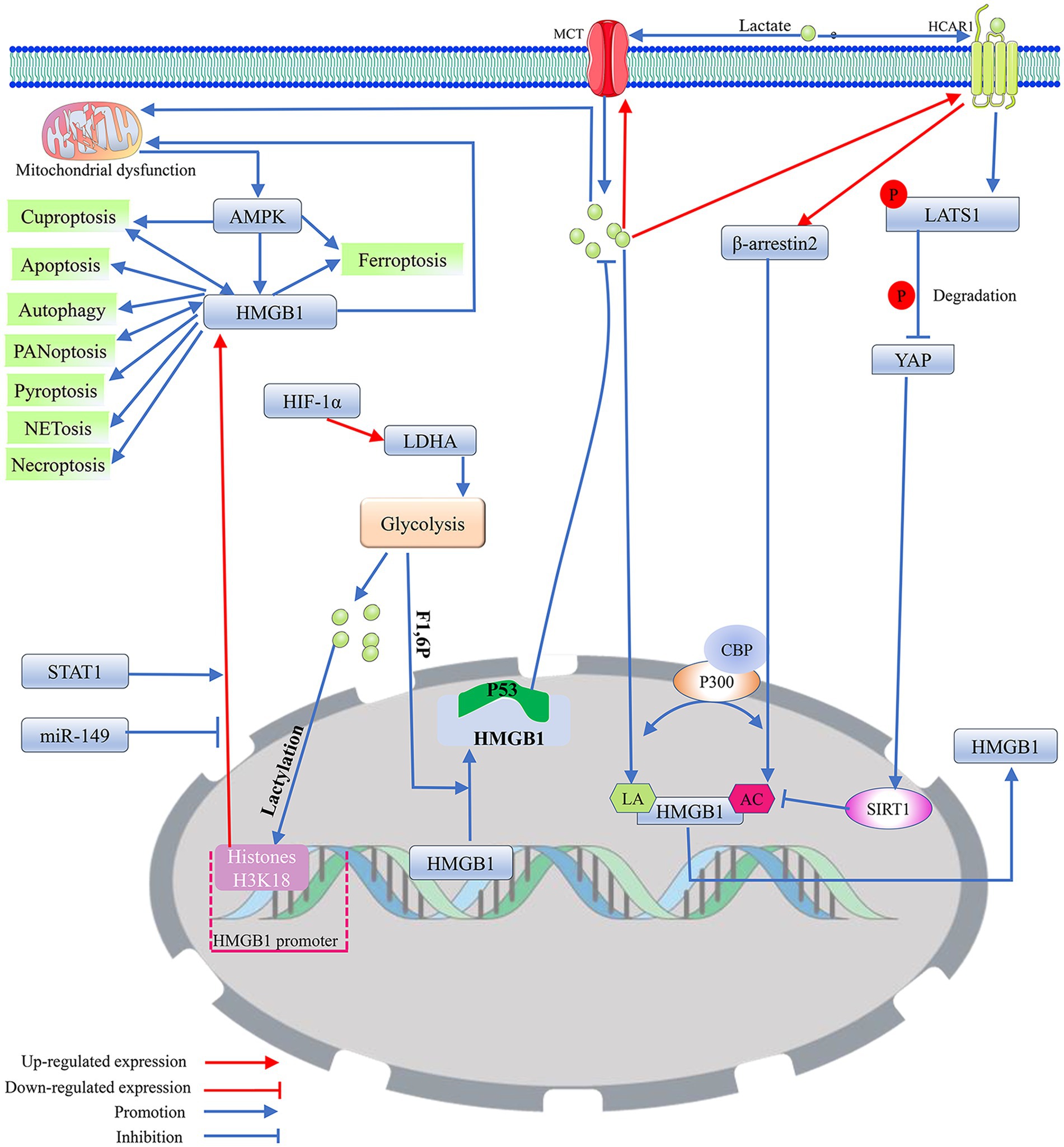
Figure 2. The potential regulatory mechanism of lactate on the biological activity of HMGB1. 1. As a DNA-binding protein, HMGB1 is usually located within the cell nucleus, but when it separates from the DNA strand and migrates out of the cell nucleus, its properties that enhance inflammation and immune responses are activated. Mitochondrial dysfunction promotes the nuclear translocation of HMGB1 through the activation of AMPK, thereby inducing HMGB1 mediated programmed cell death. Among them, cuproptosis and PANoptosis can further promote the nuclear translocation of HMGB1. In addition, miR-149 and STAT1 can affect HMGB1-mediated programmed cell death and mitochondrial dysfunction by regulating the expression level of HMGB1. 2. Exogenous lactate can upregulate β-arresin2 expression by binding to HCAR1, thereby promoting P300/CBP recruitment to the nucleus. P300/CBP promotes HMGB1 secretion and activation by catalyzing HMGB1 lactylation/acetylation. Meanwhile, the binding of exogenous lactate to HCAR1 can upregulate the level of LATS1 phosphorylation, which in turn promotes the phosphorylation and degradation of YAP. The degradation of YAP could promote the acetylation of HMGB1 by reducing SIRT1 activity. In addition, exogenous lactate can be taken up by cells via MCT, thereby mediating mitochondrial dysfunction by promoting protein lactylation, and mitochondrial dysfunction can promote HMGB1 activation. Elevated intracellular lactate levels upregulate the expression of MCT1 and HCAR1. 3. Transcription factor HIF-1α can promote glycolysis by enhancing the expression of LDHA. The lactate produced by glycolysis can not only directly promote HMGB1 lactylation but also enhance the expression of HMGB1 by upregulating the lactylation level of histone H3K18 in its promoter region. F1,6P, as an intermediate product of glycolysis, can bind to the K43/K44 sites of the HMGB1 protein to promote the separation of HMGB1 from DNA in the cell nucleus. The free form of HMGB1 can block P53 degradation by binding to the oncogenic factor P53, thereby reducing the production of lactate. Acetylation (AC); adenosine 5′-monophosphate-activated protein kinase (AMPK); CREB-binding protein (CBP); fructose-1,6-bisphosphate (F1,6P); hypoxia-inducible factor 1 (HIF-1); high-mobility group box 1 (HMGB1); hydroxy-carboxylic acid receptor 1 (HCAR1); lactylation (LA); large tumor suppressor kinase 1 (LATS1); lactate dehydrogenase A (LDHA); monocarboxylate transporter (MCT); signal transducer and activator of transcription 1 (STAT1); silencing regulatory protein 1 (SIRT1); and yes-associated protein (YAP).
2.3.3 Ferroptosis and disulfidptosisMitochondria, as the center of cellular energy metabolism, are not only the main target of cuproptosis but also the main site of iron utilization. The accumulation of excessive iron (Fe2+) in mitochondria, which is a key factor in triggering ferroptosis, can lead to mitochondrial functional impairment, which exacerbates oxidative stress-induced cellular damage through the release of ROS, destructive lipids, mitochondrial DNA, and proteins (Moos et al., 2023). Ferroptosis is a form of programmed cell death caused by lipid peroxidation associated with energy metabolism (Zhao et al., 2020). The onset and progression of ferroptosis may involve disruption of mitochondrial function, which often induces the Warburg effect (Yang et al., 2023). Elevated lactate concentration in cells may regulate iron ion metabolism by promoting lactylation modification of proteins, thereby promoting cellular ferroptosis (Zhang et al., 2023). Nevertheless, exogenous lactate can assist cells in replenishing their energy resources by activating HCAR1 or MCT1, increasing ATP production. With the enhancement of ATP levels, the potency of AMPK is gradually reduced, resulting in the inhibition of ferroptosis or cuproptosis (Xue et al., 2023) (Figure 2). Mitochondrial dysfunction is not the sole factor inducing the Warburg effect in neurons. In HT22 cells, the activation of small-conductance calcium-activated K+ (SK) channels may initiate aerobic glycolysis as the main energy source for the cell and reduce the generation of ROS during the process of mitochondrial energy metabolism to inhibit ROS-induced cellular oxidative stress, thereby preventing the cell from entering ferroptosis (Krabbendam et al., 2020).
With the continuous deepening of research on iron metabolism and ferroptosis in recent years, inhibiting ferroptosis in neuronal cells is considered a potential new strategy for treating epilepsy (Moos et al., 2023; Chen et al., 2020). It has been shown that the neuronal excitotoxicity inducer glutamate can promote ferroptosis by releasing mitochondrial ROS (Krabbendam et al., 2020). Cerebral infarction-induced cerebral ischemia and hypoxia promote the onset of glycolysis and lactate production in neuronal cells by upregulating the expression of LDHA (Yao and Li, 2023). Elevated lactate levels promote lactylation modification of lymphocyte cytoplasmic protein 1 in neuronal cells of Middle cerebral artery occlusion (MCAO) rats, thereby exacerbating cerebral infarction-induced neuronal loss, which is an important cause of post-stroke epilepsy (Zhang et al., 2023).
The neuronal solute carrier family 7 member 11 (SLC7A11)/GPX4 pathway may play a role in reducing the occurrence of epilepsy by inhibiting ferroptosis (Liang et al., 2023; Yuan et al., 2021). SLC7A11, a transport protein that facilitates the uptake of cystine and the efflux of glutamate, is instrumental in activating GPX4. This activation occurs through its role in cystine uptake, essential for the synthesis of glutathione, a key antioxidant that GPX4 uses to prevent lipid peroxidation and, consequently, ferroptosis in neuronal cells (Yang et al., 2023). However, the accumulation of disulfides, such as cystine, in the cell causes disulfide bond stress, which mediates the formation of disulfide bonds between actin cytoskeletal proteins and the breakdown of the actin filament (F-actin) network, ultimately leading to disulfidptosis (Liu et al., 2023). Therefore, the intake of cystine mediated by SLC7A11 can suppress neuronal ferroptosis while also triggering neuronal disulfidptosis. The efficiency of SLC7A11 in transporting cystine is affected by the level of glutamate in the cell. Although HIF-1α promotes the uptake of cysteine mediated by SLC7A11 by upregulating the expression of SLC1A1, lactate may not be involved in the resistance to ferroptosis driven by the glutamate metabolism of SLC7A11 (Yang et al., 2023).
2.3.4 ApoptosisRecent research has shown that neuronal autophagy and apoptosis related to epilepsy are mediated by the mTOR signaling pathway (Liu et al., 2022), and these two forms of programmed cell death may exist in a complex interplay in the pathological changes leading to neuronal loss (Arab et al., 2023). Caspase-3 is one of the key effector molecules in executing cellular apoptosis. Activated caspase-3 and DNA-binding protein HMGB1 can rapidly transfer to mitochondria and degrade mitochondrial proteins, thereby mediating the loss of hippocampal CA1 and GABAergic interneurons to maintain sustained epilepsy (Kim et al., 2021). It is evident that HMGB1, like caspase-3, can mediate neuronal apoptosis-associated epilepsy by affecting mitochondrial structure and function. Recent studies have suggested that HMGB1 activation is regulated by lactylation modifications and that this process is closely linked to cellular lactate production (Yao and Li, 2023). Lactate promotes mitochondrial hyperfission by mediating an increase in lactylation of the mitochondrial Fission 1 protein (Fis1) lysine 20 (Fis1 K20la), which can contribute to mitochondrial hyperfission by inducing ATP depletion, mitochondrial ROS overproduction, and mitochondrial damage-mediated apoptosis. Conversely, activation of PDH downregulates lactylation of Fis1 K20 by decreasing the level of lactate produced by glycolysis, thereby alleviating the apoptosis-inducing effect of mitochondrial damage (An et al., 2023). Meanwhile, inhibiting H3 histone lactylation at H3K9 and H3K14 sites can induce apoptosis (Xu et al., 2023). It follows that lactate may mediate mitochondrial function-related apoptosis by regulating lactylation modifications of histones and non-histone proteins.
2.3.5 PyroptosisPyroptosis, as an inflammatory mode of programmed cell death, can mediate the expression and release of caspase/Gasdermin D (GSDMD)-associated inflammatory factors through the activation of inflammasomes (Xia et al., 2021). The Nucleotide-binding oligomerization domain-like receptor family card domain-containing protein (NLRP3) inflammasome is an important activator of pyroptosis related to epilepsy, and it has been demonstrated that STAT3 can promote the expression of NLRP3 by catalyzing the acetylation of histone H3K9 on the NLRP3 promoter, thereby activating the NLRP3/caspase-1 pathway and exacerbating the damage to neurons in epileptic mice (Jiang et al., 2021). Epilepsy-related neuronal pyroptosis is initiated by the accumulation of ROS caused by mitochondrial impairment in neurons (Xu et al., 2023). Mitochondrial dysfunction often forces cells to obtain energy through glycolysis. A study on brain ischemia found that lactate upregulates HMGB1 expression by promoting histone lactylation of the HMGB1 promoter, which may lead to neuronal pyroptosis (Yao and Li, 2023). It is closely related to the activation of the NLRP3 inflammasome by HMGB1 (Liu et al., 2022). Thus, lactate may affect the occurrence of epilepsy by regulating neuronal apoptosis.
2.3.6 AutophagyAutophagy is a process by which cells degrade their own excess or aging organelles and misfolded proteins (Yang et al., 2020). Lactate and pyruvate can activate mitochondrial autophagy and autophagy in primary neurons and astrocytes by lowering intracellular hydrogen ion concentration at non-toxic concentrations, which facilitates the restoration of mitochondrial function for the protection of these cells from apoptosis and necrosis (Fedotova et al., 2021). It has been shown that a lack of neuronal autophagy contributes to epilepsy and that seizures may be further exacerbated by triggering autophagy dysfunction, thus creating a vicious cycle (Ali et al., 2023). Lactate not only protects neurons by regulating mitochondrial autophagy within the cells but also maintains neuronal activity by regulating mitochondrial autophagy in glial cells (Zhu et al., 2022). Moreover, lactate may further promote cytoplasmic acidification by inducing a shift in cellular energy metabolism from oxidative phosphorylation to glycolysis, thereby sustaining mitochondrial autophagy (Komilova et al., 2021). With the exacerbation of neuronal autophagy, the autophagy articulation protein Sequestosome 1 is heavily depleted, which can inhibit the activation of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/mTOR pathway. Decreased mTOR activity impedes the downregulation of the transcription factor Hif-1α, resulting in increased expression of key glycolytic enzymes LDHA and hexokinase-2 (HK2) (Song et al., 2021). In addition, SIRT1, located in the nucleus and cytoplasm, directly or indirectly promotes the formation of autophagic vesicles and improves mitochondrial function, thus blocking epileptogenesis and development (Ali et al., 2023). Because both SIRT1 and lactate can mediate autophagy in neurons to control epileptogenesis, it is likely that there is an association between SIRT1 and lactate metabolism. A recent study has confirmed that lactate can improve long-term cognitive impairment in newborns repeatedly exposed to sevoflurane by activating SIRT1-mediated hippocampal neurogenesis and synaptic remodeling (Qiu et al., 2023).
2.3.7 NecroptosisNecroptosis, which is a novel pattern of cell death associated with inflammation, is considered one of the complex mechanisms of neuronal death after status epilepticus (Roh et al., 2023). The accumulation of ROS due to mitochondrial damage can promote necroptosis of neurons in the hippocampus of acute epilepsy patients through activation of the RIPK1/RIPK3/MLKL pathway, and mitochondrial enzyme SIRT3 and the indole-derived small molecule NecroX-7 can alleviate seizures by suppressing the production of mitochondrial ROS (Roh et al., 2023; Song et al., 2021). This shows that mitochondria are important organelles that mediate epilepsy-related neuronal necroptosis. The glycolytic product lactate may regulate the activity of HIF-1α to block the release of mitochondrial ROS mediated by PDH, thereby regulating the necroptosis induced by RIPK (Chen et al., 2023; Jiang et al., 2021; Icard et al., 2023; Wong Fok Lung et al., 2020; Weindel et al., 2022; Luo et al., 2022). Furthermore, necroptosis is activated in hippocampal astrocytes and microglia to mediate neuroinflammation 4 h after status epilepticus induction (Wu et al., 2021).
2.3.8 PANoptosisPANoptosis, which is an inflammatory programmed cell death driven by PANoptosome, has key features of pyroptosis, ferroptosis, apoptosis, and necroptosis (Lin et al., 2022; González-Rodríguez and Fernández-López, 2023). PANoptosis may be present in brain tissue I/R and is a new way of causing neuronal loss (González-Rodríguez and Fernández-López, 2023). Mitochondrial dysfunction likely affects PANoptosis of neurons, as it does most programmed forms of cell death (She et al., 2023). OGD/R induces PANoptosis of neurons by inducing mitochondrial fission and dysfunction (Zeng et al., 2023). It may be related to the hypoxia-induced enhancement of glycolytic metabolism, as several enzymes involved in glycolytic metabolism have been shown to directly regulate mitochondrial functions (Liu et al., 2023). Thus, mitochondrial oxidative stress and leakage of mitochondrial contents induced by multiple factors are important triggers of PANoptosis (Zeng et al., 2023; Liu et al., 2023; Bi et al., 2022). Numerous studies have demonstrated that the structural proteins of the PANoptosome are able to induce epilepsy-associated neuronal loss by mediating crosstalk in the neuronal programmed death pathway (Xia et al., 2023; Shi et al., 2023). PANoptosis may possess characteristics of various programmed cell deaths, including but not limited to pyroptosis, apoptosis, and necroptosis (Lin et al., 2022; González-Rodríguez and Fernández-López, 2023). Although it cannot currently be confirmed that PANoptosis includes cuproptosis, the evidence suggesting that (Fe-S) cluster damage can lead to cuproptosis, ferroptosis, and PANoptosis implies that these three programmed cell death pathways may be closely related to (Fe-S) cluster-mediated mitochondrial dysfunction (Chen et al., 2023; Lin et al., 2022). Therefore, PANoptosis, which possesses characteristics of all these programmed cell death types, may also be regulated by lactate metabolism, such as pyroptosis, apoptosis, and necroptosis.
2.4 Mitochondrial dysfunction intervention in the context of epilepsy-related neuronal programmed deathAccumulating evidence indicates that the occurrence and development of epilepsy are closely related to mitochondrial dysfunction (Moos et al., 2023). Damaged mitochondria can mediate many forms of neuronal programmed cell death by releasing mitochondrial contents, such as ROS, mitochondrial DNA, and mitochondria-associated proteins, leading to epilepsy-associated neuronal cell loss (Jia et al., 2023). Mitochondria can play an anticonvulsant and anti-neuroinflammatory role by synthesizing D-fructose 1,6-diphosphate, a glycolysis intermediate that inhibits sugar metabolism (Jia et al., 2023). It has been demonstrated that artificial mitochondrial transplantation may inhibit the loss of hippocampal neurons and the activation of glial cells by ameliorating the metabolic imbalance induced by mitochondrial dysfunction, thereby reducing hippocampal damage after seizures and ameliorating epilepsy-associated psychiatric and cognitive disorders (Jia et al., 2023). Therapeutic interventions aimed at improving mitochondrial function in the central nervous system may effectively alleviate neurological damage and cognitive impairments related to epilepsy. A recent study has demonstrated that honokiol attenuates mitochondrial dysfunction and its associated oxidative stress by regulating mitochondrial ROS homeostasis and that it may stabilize mitochondrial function through activation of mitochondrial DNA transcription mediated by the glutamate receptor N-methyl-D-aspartate receptor/AMPK/peroxisome proliferator-activated receptor γ-coactivator-1α/SIRT3 pathway (Wang et al., 2022). Overexpression of SIRT3 in neuronal cells is protective against ischemia–reperfusion injury in the mouse spinal cord (Gu et al., 2023). Meanwhile, the ketogenic diet-induced overexpression of SIRT1 can maintain the normal physiological function of neurons by promoting neuronal autophagy to eliminate protein aggregates and damaged mitochondria (Dyńka et al., 2022).
Brain injury and energy deficiency promote the transport of energy substrates, such as ketones, lactate, and acetoacetate, into cells by upregulating the number of MCT channels on the neuronal membrane, thereby ensuring normal neuronal function. Astrocytes may reduce glucose availability and frequency of epileptic seizures by producing ketones and shuttling them via MCT channels to neurons (Dyńka et al., 2022). However, some other studies suggest that transporting these energy substances and mitochondria may increase neuronal excitability, thereby triggering epilepsy. The energy substances and mitochondria transferred from glial cells to neurons may lead to neuronal excitation, an important factor in inducing epilepsy (Nagy et al., 2018; Wei et al., 2023; Jia et al., 2023). The increased synthesis of lactate caused by cerebral ischemia promotes mitochondrial apoptotic pathway-mediated neuronal death, which may induce the occurrence of post-stroke epilepsy by upregulating the lactylation levels of key proteins in the calcium signaling pathway, such as Scl25a4 (K245), Slc25a5 (K96), and Vdac1 (K20, K266) (Yao et al., 2023). However, when lactate and ketones replace glucose as the main energy source for neurons, glucose can enter the pentose phosphate pathway and produce a large amount of nicotinamide adenine dinucleotide phosphate, which can offset the oxidative effect of ROS released during the mitochondrial utilization of lactate for production (Mayorga-Weber et al., 2022). During status epilepticus, HMGB1, a non-histone DNA-binding protein, can be transferred from the cell nucleus to the mitochondria, thereby disrupting the energy metabolism balance of the mitochondria (Kim et al., 2021). HMGB1 in microglia can promote the release of inflammatory factors such as IL-1β and IL-18 by activating the NLRP3/NF-κB/MAPKs pathway, leading to the activation of the NLRP3 inflammasome in hippocampal neurons, which can subsequently lead to pyroptosis and apoptosis (Liu et al., 2022; Shen et al., 2018). The neuronal injury promotes the release of neurally mediated ATP, which reactivates the NLRP3/NF-κB/MAPKs pathway via binding to the purinergic receptor P2X ligand-gated ion channel 7 in microglia without HMGB1. HMGB1 may be a central element in the tandem of neuronal death and glial cell activation in the hippocampus, and HMGB1 can amplify the effects of neuroinflammatory injury by promoting the interaction between neuronal death and glial cell activation (Liu et al., 2022; Shen et al., 2018) (Figure 1).
2.5 Regulation of neuronal programmed death by HMGB1 2.5.1 HMGB1 and mitochondriaHMGB1 nucleus-to-cytoplasm translocation has been shown to maintain status epilepticus and drug-resistant temporal lobe epilepsy by mediating oxidative stress in brain tissue in neurons and glial cells (Pauletti et al., 2019). The function and activity of HMGB1 are determined not only by its subcellular localization but also by its oxidation–reduction status and post-translational modifications (Chen et al., 2023). Mitochondrial damage-mediated oxidative modification of HMGB1 promotes cytoplasmic accumulation and extracellular secretion of HMGB1. HMGB1 secreted into the extracellular compartment initiates the expression of NF-κB-mediated inflammatory factors (e.g., TNF-α and IL-1β) through activation of Toll-like receptor 4 (TLR4) on the cell surface, ultimately leading to inflammatory tissue damage (Zhao et al., 2023). Brain tissue ischemia induces HMGB1 translocation from the cell nucleus to the mitochondria and extracellular region in neurons and astrocytes, leading to neuronal apoptosis associated with epileptic status (Wang et al., 2020; Dai et al., 2023). During this process, the transposition of HMGB1 triggers the destruction of mitochondria, and the released contents of mitochondrial lysis further intensify HMGB1-mediated neuroinflammatory damage (Wang et al., 2020; Dai et al., 2023; Zhang et al., 2020) (Figure 2).
2.5.2 HMGB1 and various types of programmed cell deathMitochondrial dysfunction can induce epileptogenesis by mediating multiple programmed cell deaths in neurons, and HMGB1 may intervene in epilepsy-associated multiple programmed cell deaths by modulating mitochondrial function. It has been demonstrated that HMGB1 released into the extracellular space can act as a damage-associated molecular pattern protein to mediate a variety of cellular programmed deaths (Chen et al., 2023; Tang et al., 2023). HMGB1, which is secreted extracellularly, may drive the disease process in epilepsy by inducing neuroinflammation and neuronal loss during the silent phase following the initial triggering event (Rosciszewski et al., 2019). HMGB1 transferred from the nucleus to the cytoplasm may activate the Myeloid differentiation factor 88 (MyD88)/NF-κB axis by binding to TLR4. The MyD88/NF-κB axis can induce neurological deficits by promoting neuronal apoptosis, autophagy, and the release of inflammatory factors, such as IL-1β and TNF-α (Lei et al., 2022; Fu et al., 2023; Shang et al., 2020) (Figure 3). STAT1 may upregulate the expression level of HMGB1 by binding to the HMGB1 promoter, thereby affecting the biological effects mediated by the TLR4/MyD88/NF-κB pathway (Fu et al., 2023). In the MCAO model, impaired neurons induce NETosis by releasing large amounts of HMGB1, which induces NETosis and can even be extruded as part of the NET. The extruded HMGB1 may further induce NETosis and platelet activation, which is one of the important reasons for the elevation of HMGB1, thereby exacerbating neuronal inflammatory damage (Kim and Lee, 2020). miR-149 may reduce the occurrence of epilepsy by inhibiting HMGB1-mediated neuroinflammatory injury (Kwak et al., 2023; Parsons et al., 2022; Wang et al., 2022). Cuproptosis and PANoptosis may further induce the release of inflammatory factors and programmed cell death by promoting the release of HMGB1 (Liu et al., 2022; Kwak et al., 2023). In addition, ATP depletion and calcium ions can upregulate the level of HMGB1 phosphorylation through activation of AMPK, which can promote the translocation and activation of HMGB1 (Liu et al., 2022). It cannot be excluded that HMGB1 may induce various forms of programmed death in neurons by regulating mitochondrial function (Figure 2).
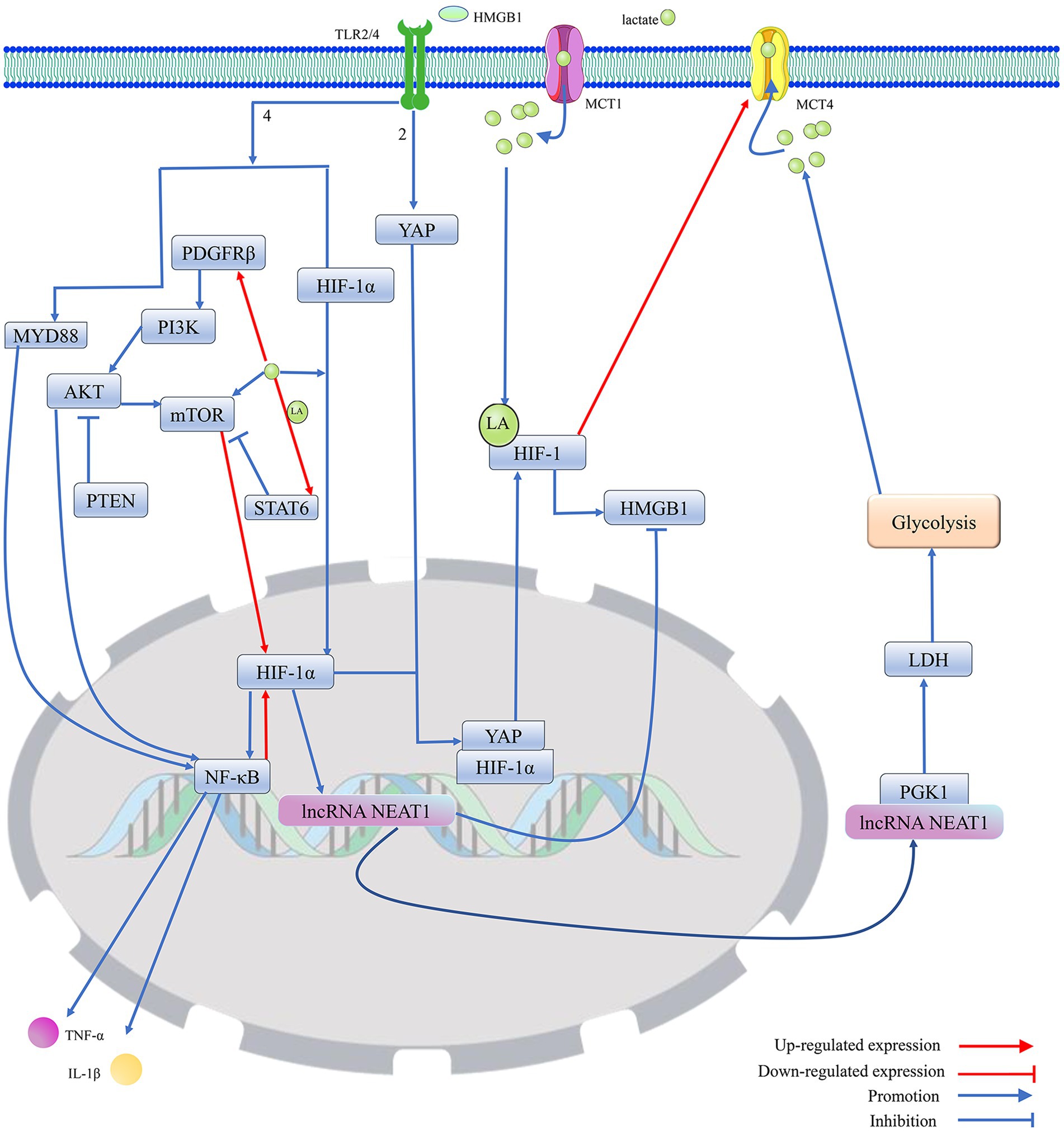
Figure 3. Protein lactonylation modification is an important influence in the process that is HIF-1-mediated HMGB1 activity. 1. Lactate, as a metabolite of cellular adaptation to hypoxic environments, is mainly produced by glycolysis. MCT1 and MCT4 are mainly responsible for the cellular uptake and release of lactate. Elevated intracellular lactate levels not only promote the translocation of HIF-1α from the cytoplasm to the nucleus to regulate gene expression but also stabilize the structure and enhance the biological activity of HIF-1 by promoting HIF-1 lactylation. Furthermore, lactate can promote the expression of HIF-1α by directly or indirectly enhancing the activation of mTOR. However, lactate can upregulate STAT6 expression by promoting histone lactylation, thereby blocking PDGFRβ/PI3K/AKT/mTOR-mediated HIF-1 expression. 2. Activated HIF-1 promotes the secretion and activation of HMGB1, which is secreted into the extracellular compartment and promotes the migration of YAP from the cytoplasm to the nucleus by binding to TLR2, thereby promoting the activation of HIF-1. Meanwhile, HMGB1, by binding to TLR4, not only directly promotes the migration of HIF-1 to the nucleus to exert transcriptional regulation but also activates the MYD88/NF-κB pathway to enhance the expression of HIF-1. However, HIF-1α can reduce the expression of HMGB1 by upregulating the transcript level of lncRNA NEAT1, thereby blocking the excessive activation of HIF-1 induced by HMGB1. The lncRNA NEAT1 can stabilize the structure of PGK1 by binding to PGK1, which mediates the glycolysis/lactate/HIF-1α pathway by increasing LDH activity, but HIF-1 can promote lactate efflux by upregulating the expression of MCT4, which blocks the excessive activation of HIF-1 induced by lactate. Furthermore, PTEN could block HIF-1 expression, which is mediated by the PDGFRβ/PI3K/AKT/NF-κB pathway, by inhibiting AKT, thereby reducing HMGB1-induced release of cellular inflammatory factors. Protein kinase B (AKT); hypoxia-inducible factor 1 (HIF-1); high-mobility group box. 1 (HMGB1); interleukin 1β (IL-1β); lactylation (LA); lactate dehydrogenase (LDH); monocarboxylate transporter (MCT); mechanistic target of rapamycin (mTOR); myeloid differentiation factor 88 (MYD88); nuclear enriched transcript 1 (NEAT1); platelet-derived growth factor receptor β (PDGFRβ); phosphoglycerate kinase 1 (PGK1); phosphatidylinositol 3-kinase (PI3K); phosphatase and tensin homolog (PTEN); signal transducer and activator of transcription 6 (STAT6); toll-like receptor (TLR); tumor necrosis factor-α (TNF-α); and yes-associated protein (YAP).
Neuroglia may be involved in HMGB1-mediated neuronal programmed cell death (Rosciszewski et al., 2019). HMGB1 induces NF-κB pathway activation in glial cells by activating signaling pathways involving TLR2, TLR4, and the receptor for advanced glycation end products (RAGE). Since activation of the NLRP3/NF-κB/MAPKs pathway in microglia is an important mechanism for HMGB1-induced hippocampal neuronal pyroptosis and apoptosis (Liu et al., 2022; Shen et al., 2018), the limited activation of NF-κB in astrocytes can lead to partial suppression of neuron loss in the absence of microglia, thereby producing a beneficial effect on controlling the occurrence of epileptic seizures (Cai and Lin, 2022; Rosciszewski et al., 2019). The damaged neurons can further promote the activation of microglia by secreting HMGB1 (Kitaoka et al., 2023). HMGB1 secreted by neurons can also directly act on astrocytes, thereby activating NF-κB p65-mediated inflammatory cascade effects (Kaya et al., 2023). It can be speculated that the intervention of glial cells may exacerbate the neuronal death effect induced by HMGB1.
2.5.3 The impact of lactate metabolism on the function of HMGB1Research has shown that the increased protein lactylation level of hippocampal neurons is accompanied by upregulation of the HMGB1 expression level (Xie et al., 2023). Extracellular lactate can regulate cell death and immune cell polarization by binding to HCAR1 and transp
留言 (0)