
記住我
Primary coenzyme Q10 (CoQ10) deficiency is a heterogeneous genetic disorder caused by mitochondrial dysfunction. It is an autosomal recessive disease that most commonly manifests in childhood and may involve the nervous system, kidney, skeletal muscle and heart. When the kidney is involved, the typical presentation is steroid-resistant nephrotic syndrome (SRNS).
CoQ10 is essential for the proper functioning of the mitochondrial respiratory chain. CoQ10 is essential for cellular metabolism and energetics with crucial roles in ATP production, pyrimidine biosynthesis and the regulation of apoptosis. There are limited reports describing the pathogenic effects of COQ8B variants on CoQ biosynthesis and other related mitochondrial functions, especially in children with kidney disease. Insights from structural modeling suggest that pathogenic variants in COQ8B alter allosteric regulation or protein folding and stability in renal disease (1). A family of at least 17 different genes synthesize CoQ10 in mitochondria (2). COQ8-related genes include COQ8A (NM_020247.5) and COQ8B (NM_024876.4). A recent study showed that approximately 5.8% of SRNS patients had biallelic COQ8B variants. More than 26 variants have been reported in COQ8B alone according to the Human Gene Mutation Database (HGMD; https://www.hgmd.cf.ac.uk/ac/index.php). Clinically, COQ8B - related glomerulopathy presents as SRNS in childhood most commonly without associated extrarenal manifestations. FSGS is the most common histopathologic finding and the response to therapy can be heterogeneous. Here, we report the clinical features and therapeutic responses of 4 pediatric patients with glomerular disease due to COQ8B variants.
Case presentationsA total of 4 patients (2 females and 2 males) from 2 unrelated families were included in this study.
Family 1The oldest of three affected siblings from a 3-generation kindred was diagnosed with non-syndromic proteinuric kidney disease at age 9-year old (Figure 1a). She died two years later from kidney failure and had not received CoQ10. Her two surviving siblings who had proteinuria and normal renal function, underwent clinical exome sequencing and genotyped for a homozygous COQ8B variant (c.737G>A) (Figure 2a). The family denies a history of consanquinity and no we were not able to identify evidence of consanguineous relationships through our analyses. Based on these findings, oral CoQ10 therapy was initiated. CoQ10 treatment was effective in reducing proteinuria over the first six months of therapy and the siblings have retained normal renal function (Table 1).
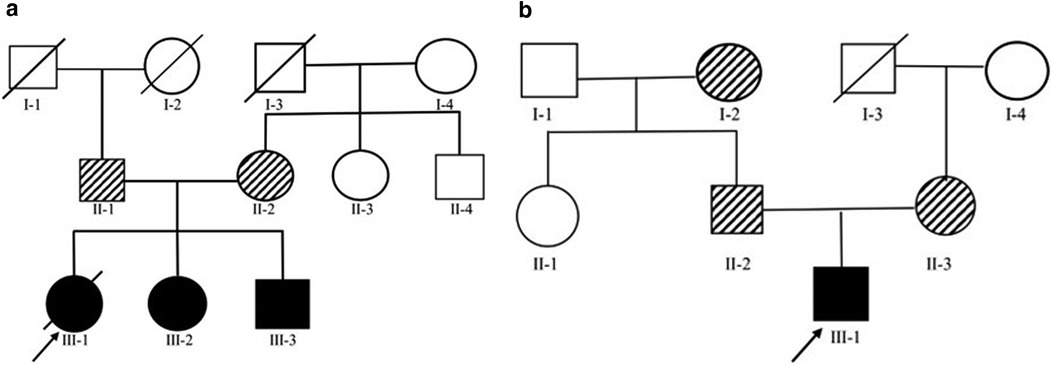
Figure 1. (A) Family 1 Pedigree - Cases 2 and 3 both carried homozygous variants of COQ8B with in exon 9 (c.737G>A) while both parents heterozygously carried the same variant. (B) Family 2 Pedigree - Case 4 carried two heterozygous variants, c.737G>A (a) and c.1465C>T (d) in COQ8B. While his mother and father heterozygously carried c.737G>A (c) and c.1465C>T (e), respectively.
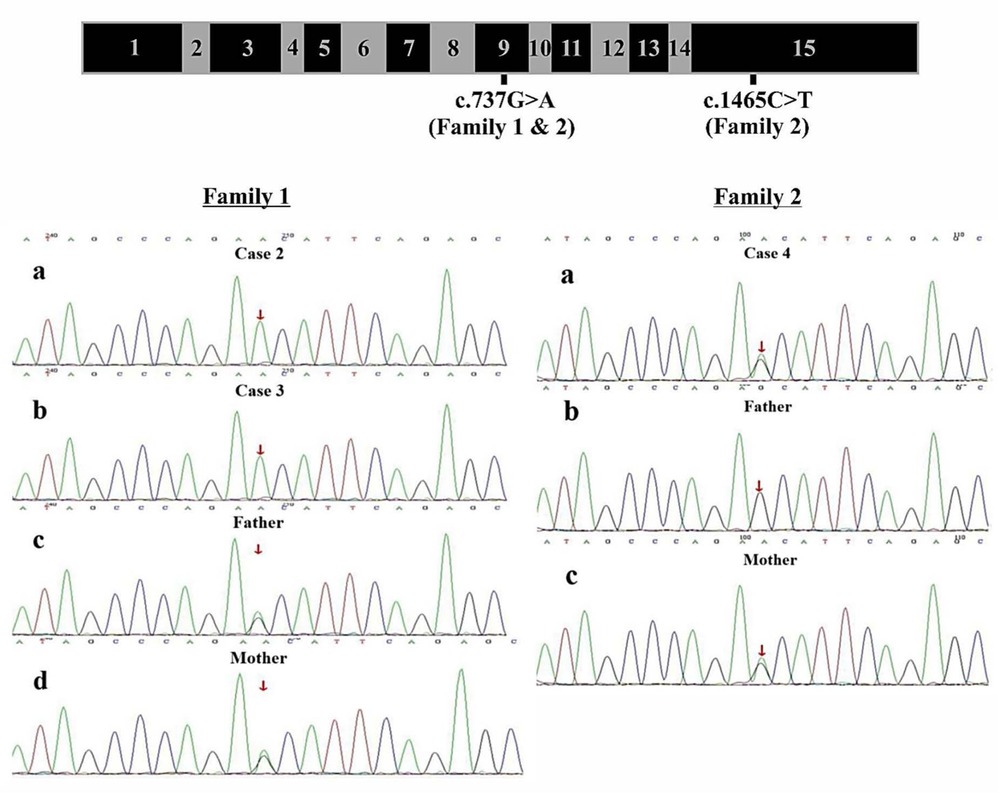
Figure 2. Genomic Structure of COQ8B and Family Sequencing -COQ8B gene structure with labeling of family variants in exons 9 and 15. Direct sequencing data for Family 1 showing 2 affected siblings with the homozygous COQ8B variant (c.737G>A) and both unaffected parents with a heterozygous COQ8B variants. Direct sequencing data for Family 2 showing 1 affected individual with a compound heterozygous variants (c.737G>A and c.1465C>T) of COQ8B and each unaffected parent with a single heterozygous variant.
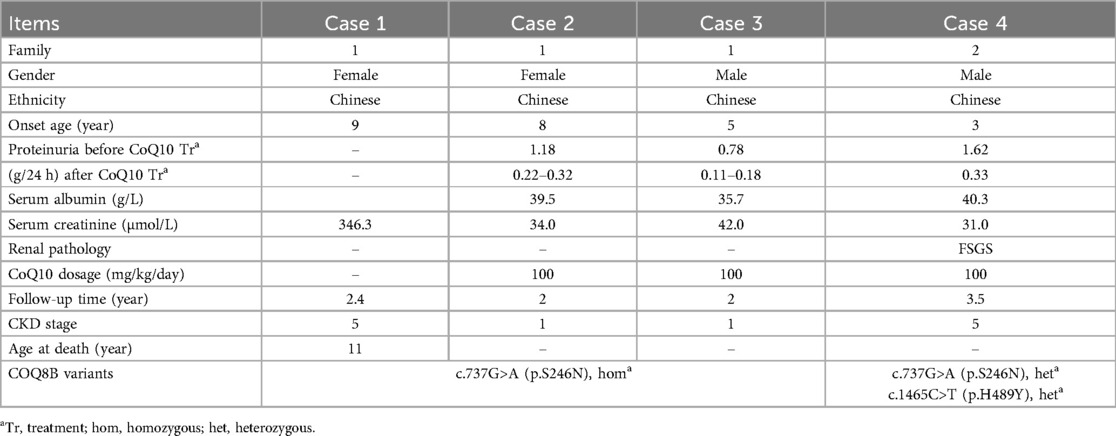
Table 1. Clinical data for the families 1 & 2.
Family 2A fourth individual was diagnosed with FSGS (Figure 1b). The patient was initially treated with glucocorticoids and cyclosporine without response. Clinical exome analyses revealed a pathogenic compound heterozygous variant of COQ8B (c.737G>A and c.1465C>T) (Figure 1b). Based on this finding, oral CoQ10 therapy was initiated. CoQ10 was effective in reducing his proteinuria over the first ∼4 months of therapy, however, his renal function began to deteriorate after 1 year of treatment and he progressed to kidney failure (Table 1). Kidney transplantation wassubsequently performed and his condition stabilized with no recurrence of disease.
Parents of both families were healthy, with non-consanguineous marriages or family history of kidney disease. After obtaining informed consent from all parents, peripheral blood was collected from patients and their respective parents for genetic analyses. Clinical exome testing and analyses were performed by MyGenostics Laboratory in China. Results showed a homozygous variant c.737G>A (p.S246N) (PM3) of COQ8B gene within exon 9 which was reported in The Human Gene Mutation Database (HGMD; https://www.hgmd.cf.ac.uk/ac/index.php). Both parents were heterozygous for the variant (Figure 2). The expected segregation of putative variants was confirmed in families, whenever possible, and their absence was confirmed in SNPs databases of common benign variants (http://www.ncbi.nlm.nih.gov/projects/SNP/ and www.gnomad-sg.org). Human Splicing Finder (http://www.umd.be/HSF/) and Mutation Taster (http://mutationtaster.org/) were used for pathogenicity prediction of the variants, respectively andthe c.737G>A (p.S246N) variant were predicted to be “damaging” or “possibly damaging”.
In Family 2, the exome analysis of the male proband showed two heterozygous variants c.737G>A (p.S246N) and c.1465C>T (p.H489Y), in exon 9 and exon 15 of COQ8B gene, respectively (Figure 2). His mother carried the c.737G>A variant while his father carried the c.1465C>T variant (Figure 2). The rare c.737G>A variant was found in the SNP databases (rs200841458, A = 0./0, 0.000043/6 and 0.000057/15 in ALFA, GnomAD and TOPMED, both heterozygotes, respectively)with a minor allele frequency of 0.000037 and was found almost exclusively in individuals of East Asian ancestry. The c.1465C>T (p. H489Y) variant was also found in the SNP databases (rs139063940, A = 0./0, 0.000013/3 and 0.000008/2 in ALFA, GnomAD and TOPMED, both heterozygotes, respectively),was rare with a minor allele frequency of 0.0000037, and was almost exclusively expressed in individuals of East Asian ancestry. PolyPhen-2, SIFT, Variant Taster and GERP++ analysis predicted that both the two variants were “damaging” or “possibly damaging”. Additionally, both the p.S246N and the p.H489Y variants are predicted to disrupt the secondary structure of the COQ8B protein (Figure 3).
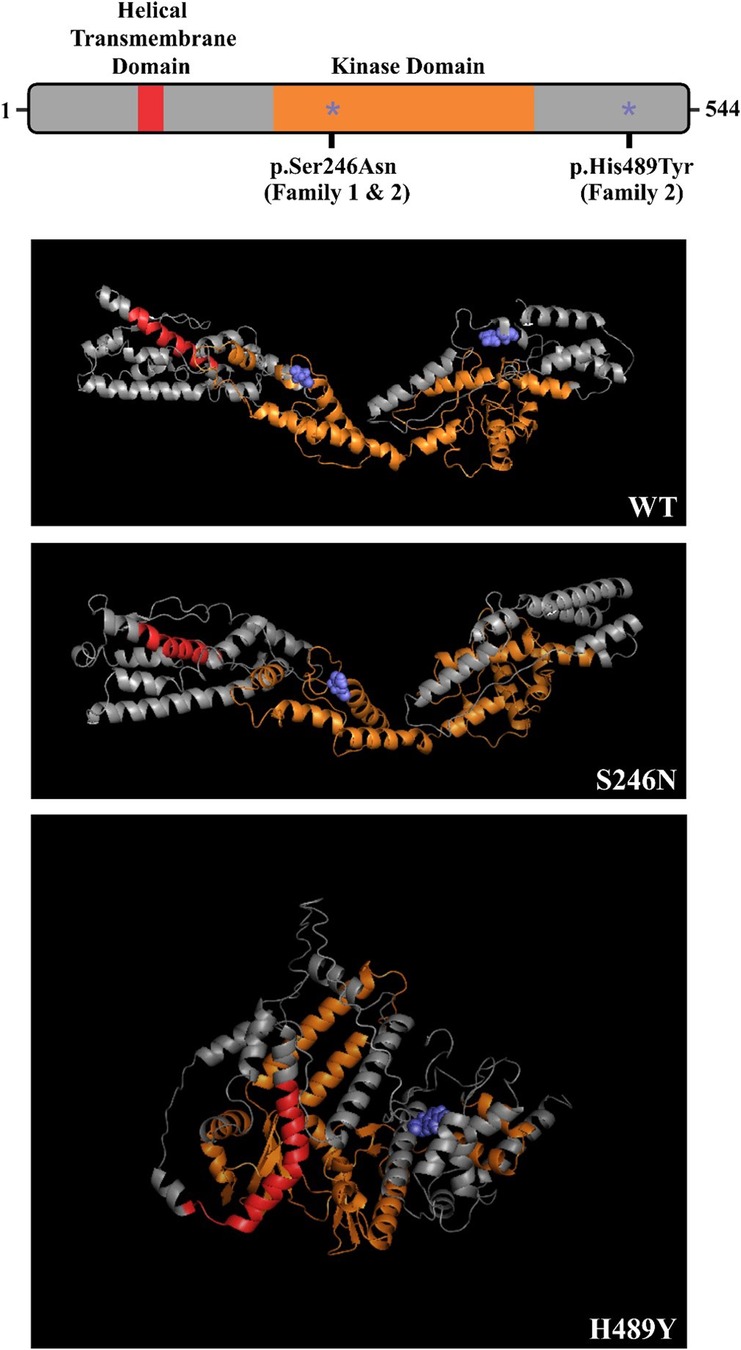
Figure 3. Predicted effect of family COQ8B variants on protein secondary structure – COQ8B domain structure depicted with p.S246N and p.H489Y variants labeled (*). Both variants are predicted to disrupt the secondary structure of the protein.
After diagnosis was confirmed as COQ8B-related glomerulopathy, cases 2, 3 and 4 were treated immediately with oral supplementation of CoQ10 (100 mg/kg/day). In case 2 from Family 1, her appetite improved, weight began to increase, and urine protein began to decrease after the first month of treatment. Her weight increased by 3 kg and her total 24-h urine protein decreased to 0.27 g after 3 months. Her total 24-h urine protein remained between 0.22 and 0.32 g, and her renal function remained normal over 2 years of follow up.
In case 3 from Family 1, the total 24-h urine protein decreased to 0.13 g after 3 months and remained between 0.11 and 0.18 g over 2 years follow up. His renal function has remained normal.
In case 4 from Family 2, his total 24-h urine protein decreased to 0.33 g after ∼4 months of CoQ10 supplementation. However, his proteinuria and serum creatinine level began to increase after 1 year (Figures 4a,b). His kidney biopsy showed FSGS (Figure 5). Under light microscopy, Case 4 shows 43 glomeruli, 21 of which were sclerotic, and vacuolar degeneration was observed in the renal tubules. Electron microscopy findings were consistent with the light microscopy and no mitochondrial morphological abnormalities were detected. He reached CKD stage 5 two years later and peritoneal dialysis was initiated. He underwent kidney transplantation after 2.5 years. He has experienced no episodes of rejection and no recurrence of disease post-transplant. During the entire treatment period, the child had no severe or recurrent infections and was not exposed to any nephrotoxic drugs.
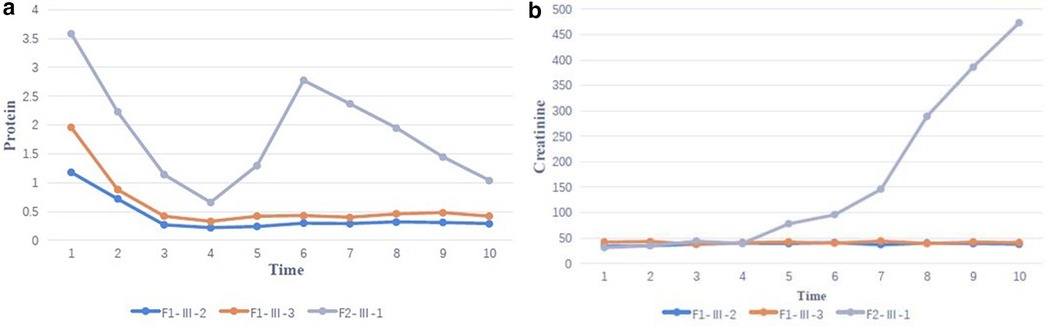
Figure 4. (a) Family proteinuria response trajectories - change in proteinuria during coenzyme Q10 (CoQ10) supplementation over 2 years of follow-up (F1-Ⅲ-2, F1-Ⅲ-3 and F2-Ⅲ-1 correspond to case 2,3 and 4). (b) Family eGFR Response Trajectories – Change in serum creatinine during coenzyme Q10 (CoQ10) supplementation over 2 years of follow-up (F1-Ⅲ-2, F1-Ⅲ-3 and F2-Ⅲ-1 correspond to case 2,3 and 4).
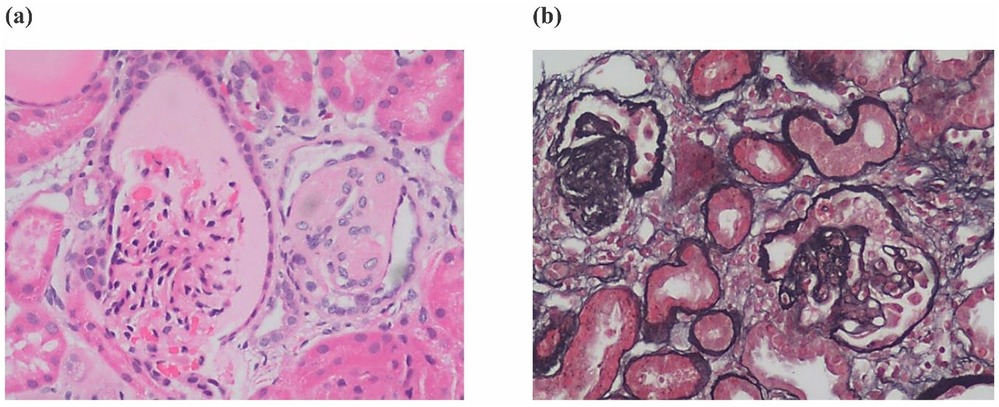
Figure 5. Renal histopathology - (a) renal histopathology of case 4 showing FSGS by H&E staining and (b) Silver Jones Methenamine staining.
DiscussionPathogenic COQ8B variants can cause primary CoQ10 deficiency; a clinically and genetically heterogeneous disorder. COQ8B gene variant-related glomerulopathy is a recently recognized glomerular disease associated with coenzyme Q deficiency that most commonly manifests in early childhood as SRNS with FSGS histopathology that can progress to kidney failure (1, 2). SRNS is one of the leading causes of kidney failure in children and young adults, and its renal histopathology is most commonly FSGS (3, 4). Reports suggest that COQ8B-related glomerulopathy is a progressive disease and an important consideration in the differential diagnosis for adolescent kidney failure patients (3–6). CoQ10 deficiency often affects multiple systems, including the nervous system, kidneys, skeletal muscle, and heart. Reports of extrarenal manifestations in children with COQ8B-related glomerulopathy are rare (7). A total of 4 patients (2 females and 2 males) from 2 unrelated families were evaluated in this study. In both families, neither of the patients had extrarenal manifestations of disease and all affected individuals presented with isolated proteinuria with or without renal dysfunction. This report is unique because there have been no cases previously reported in the Han Chinese subpopulation in northern China. Additionally, the cases highlighted in this report delineate the treatment outcomes of three children who received similar care after ∼3 years of follow up. As with other cases, the reason for the heterogeneity of the treatment responses in these families is not clear. This re-emphasizes the idiosyncratic nature of genetic kidney disease and the urgent need for larger randomized controlled trials of diagnosis and therapy for monogenic COQ8B-associated nephropathy to establish optimal treatment algorithms for the disease and to define at-risk populations. Such studies are also needed to provide more precise frequency estimates of the burden of disease. In 2018, Wang et al. showed that CO8QB variants accounted for the 6.67% of monogenic SRNS cases in a large Chinese cohort (8). CO8QB variants were the most common cause of monogenic SRNS in this population suggesting a unique ancestry-driven predisposition and highlighting a need for early genetic testing in an at-risk population.
FSGS was the most common pathological finding on renal biopsy in COQ8B-related glomerulopathy (7). In this study, the renal histopathology of Case 4 was FSGS while the other 3 cases did not undergo renal biopsy. The histopathology of COQ8B-related glomerulopathy lacks pathognomonic morphological features, and the specific mechanism of its predisposition to FSGS is unknown. COQ8B is expressed in mitochondria of podocytes, proximal tubules and collecting ducts. There are no characteristic features of CoQ10 deficiency by light other than the FSGS lesion, however, several reports have documented abnormal tubular cell morphology characterized as granular swollen epithelial cells (GSECs). Therefore, identifying GESCs by light microscopy may be useful for identification of COQ8B nephropathy and other CoQ10 deficiencies (9). Renal histopathology of some patients manifested as mild glomerular abnormalities by light microscopy, together with mitochondrial proliferation, hypertrophy and crowding by electron microscopy (10). COQ8B glomerulopathy often does not respond to corticosteroid and immunosuppressive therapy. Fortunately, some patients benefit from early coenzyme Q10 supplementation. In our study, the oldest affected sibling (Case 1) from Family 1 was diagnosed with kidney failure at the age of 9 and died at 11 years old. No genetic diagnosis was established and she did not receive CoQ10. Coenzyme Q10 treatment was effective in reducing proteinuria and appeared to be protective in the other 3 cases, with a significant reduction in urinary protein after about half a year of treatment. Unfortunately, proteinuria began to increase after 1 year of follow-up in this patient as his disease progressed. The therapeutic effect of coenzyme Q10 supplementation is not uniform in patients with renal disease associated with coenzyme Q deficiency. This may due to a variety of factors such as delay in the onset of therapy, variability in disease chronicity, severity of the disease-causing genetic variant, additive or synergistic intergenic interactions, etc. Documented responses vary from no response, partial remission, or complete disappearance of urinary protein (4, 11, 12). Efficacy of supplementation of exogeneous CoQ10 has been shown in some patients with CoQ10 nephropathies and the rare side effects of treatment were mild (13). Initiating exogenous coenzyme Q10 (CoQ10) supplementation early in the asymptomatic period has been shown to reduce total urinary protein and appears to have a protective effect on renal function in some children (14). Other studies have demonstrated that CoQ10 supplementation led to significantly improved preservation of kidney function (15). Despite these cases of treatment success, other studies have shown less favorable outcomes suggesting that successful treatment may depend on a diversity of factors (i.e., genetic diagnosis, timing of therapy initiation, comorbid disease, etc.) (16). The reason for the non-uniform therapeutic effect of coenzyme Q10 supplementation remains unclear and there is no genotype-phenotype correlation described so far. Notably, there are no studies addressing whether supplementation of CoQ10 has a reno-protective potential if started in an asymptomatic period before irreversible renal damage is established. There are also no studies on the specific dose and duration of oral supplementation with CoQ10. Most physicians still recommend early application and life-long treatment. It has also been suggested that early detection of COQ8B nephropathy after supplementation with CoQ10 in combination with ACE inhibitors can slow the progression of renal insufficiency. Kidney tranplantation in patients with familial SRNS, such as COQ8-associated nephropathy, was not associated with recurrence of the underlying diseas (17). The commonly used dose is 100 mg/kg/day, which was also adopted in our treatment strategy. Of our 3 patients who received exogenous COQ10, 2 responded well to this regimen and their renal function remains normal to date. However, in case 4, renal function initially improved with treatment, but began to deteriorate 1 year later, and eventually progressed to kidney failure. This is consistent with other cases reported in the literature (18). Therefore, the protective effect of exogenous COQ10 on renal function still needs further observation and research, which may be affected by rarity of the disease. In summary, genetic testing, including investigation of genes involved in CoQ10 biosynthesis, should be considered in young patients with proteinuria of unknown etiology or CKD, especially without extrarenal manifestations. Early initiation of therapy may reduce proteinuria and preserve kidney function.
ConclusionCOQ8B-associated glomerulopathy often presents as SRNS with no obvious extrarenal manifestations. It has been identified as a significant cause of monogenic nephropathy in some populations and exhibits a variable response to CoQ10. The histopathology is predominantly FSGS, and there are no pathognomonic morphological features of the disease. Patients often do not respond to corticosteroid or immunosuppressive therapy, however, a considerable number of children may respond to early supplementation with coenzyme Q10 therapy. In these patients, CoQ10 can reduce proteinuria and may protect kidney function. Unfortunately, some patients will still develop to kidney failure. For these individuals, kidney transplantation can an effective treatment option. For children with unexplained proteinuria and abnormal renal function, genetic testing should be performed early in the course of disease as a diagnosis of COQ8B-glomerulopathy may be treatable.
We acknowledge the limitations of this concise report, and we will pursue further studies in a larger cohort in the future. Due to the limited sample size, our study results may not be generalizable to the Han Chinese population. To address these limitations, a broader evaluation of familial SRNS cases in this population is needed to better characterize the epidemiology of COQ8B-associated nephropathy and the efficacy of CoQ10 therapy. None the less, our findings are consistent with prior reports in other subgroups and identify the Han Chinese as an at-risk population for the SRNS caused by pathogenic COQ8B variants.
Data availability statementThe original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding authors.
Ethics statementThe studies involving humans were approved by Hebei Provincial Children's Hospital, China. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributionsLZ: Writing – review & editing. GH: Writing – review & editing. PH: Data curation, Formal Analysis, Writing – original draft. CL: Methodology, Writing – original draft. JC: Software, Writing – original draft.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. GH is supported by National Institutes of Health grant NIH K08-DK111940.
AcknowledgmentsThe authors thank the patients and their family members for participating in this study.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. AbuMaziad AS, Thaker TM, Tomasiak TM, Chong CC, Galindo MK, Hoyme HE. The role of novel COQ8B mutations in glomerulopathy and related kidney defects. Am J Med Genet A. (2021) 185:60–7. doi: 10.1002/ajmg.a.61909
PubMed Abstract | Crossref Full Text | Google Scholar
2. Vazquez Fonseca L, Doimo M, Calderan C, Desbats MA, Acosta MJ, Cerqua C, et al. Mutations in COQ8B (ADCK4) found in patients with steroid-resistant nephrotic syndrome alter COQ8B function. Hum Mutat. (2018) 39:406–14. doi: 10.1002/humu.23376
PubMed Abstract | Crossref Full Text | Google Scholar
3. Zhu X, Zhang Y, Yu Z, Yu L, Huang W, Sun S, et al. The clinical and genetic features in Chinese children with steroid-resistant or early-onset nephrotic syndrome: a multicenter cohort study. Front Med. (2022) 9:885178. doi: 10.3389/fmed.2022.885178
PubMed Abstract | Crossref Full Text | Google Scholar
4. Korkmaz E, Lipska-Ziętkiewicz BS, Boyer O, Gribouval O, Fourrage C, Tabatabaei M, et al. ADCK4 - associated glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol JASN. (2016) 27:63–8. doi: 10.1681/ASN.2014121240
PubMed Abstract | Crossref Full Text | Google Scholar
5. Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. (2013) 123:5179–89. doi: 10.1172/JCI69000
PubMed Abstract | Crossref Full Text | Google Scholar
6. Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol CJASN. (2018) 13:53–62. doi: 10.2215/CJN.04120417
PubMed Abstract | Crossref Full Text | Google Scholar
7. Park E, Kang HG, Choi YH, Lee KB, Moon KC, Jeong HJ, et al. Focal segmental glomerulosclerosis and medullary nephrocalcinosis in children with ADCK4 mutations. Pediatr Nephrol. (2017) 32:1547–54. doi: 10.1007/s00467-017-3657-9
PubMed Abstract | Crossref Full Text | Google Scholar
8. Wang F, Zhang Y, Mao J, Yu Z, Yi Z, Yu L, et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr Nephrol Berl Ger. (2017) 32:1181–92. doi: 10.1007/s00467-017-3590-y
PubMed Abstract | Crossref Full Text | Google Scholar
10. Maeoka Y, Doi T, Aizawa M, Miyasako K, Hirashio S, Masuda Y, et al. A case report of adult-onset COQ8B nephropathy presenting focal segmental glomerulosclerosis with granular swollen podocytes. BMC Nephrol. (2020) 21:376. doi: 10.1186/s12882-020-02040-z
PubMed Abstract | Crossref Full Text | Google Scholar
11. Zhai S, Zhang L, Sun B, Zhang Y, Ma Q. Early-onset COQ8B (ADCK4) glomerulopathy in a child with isolated proteinuria: a case report and literature review. BMC Nephrol. (2020) 21:406. doi: 10.1186/s12882-020-02038-7
PubMed Abstract | Crossref Full Text | Google Scholar
12. Feng C, Wang Q, Wang J, Liu F, Shen H, Fu H, et al. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation: case reports and literature review. Medicine (Baltimore). (2017) 96:e8880. doi: 10.1097/MD.0000000000008880
PubMed Abstract | Crossref Full Text | Google Scholar
13. Atmaca M, Gülhan B, Atayar E, Bayazıt AK, Candan C, Arıcı M, et al. Long-term follow-up results of patients with adck4 mutations who have been diagnosed in the asymptomatic period: effects of early initiation of coq10 supplementation. Turk J Pediatr. (2019) 61:657. doi: 10.24953/turkjped.2019.05.003
PubMed Abstract | Crossref Full Text | Google Scholar
14. Drovandi S, Lipska-Ziętkiewicz BS, Ozaltin F, Emma F, Gulhan B, Boyer O, et al. Oral coenzyme Q10 supplementation leads to better preservation of kidney function in steroid-resistant nephrotic syndrome due to primary coenzyme Q10 deficiency. Kidney Int. (2022) 102:604–12. doi: 10.1016/j.kint.2022.04.029
PubMed Abstract | Crossref Full Text | Google Scholar
16. Yang J, Yang Y, Hu Z. A novel ADCK4 mutation in a Chinese family with ADCK4-associated glomerulopathy. Biochem Biophys Res Commun. (2018) 506:444–9. doi: 10.1016/j.bbrc.2018.10.102
PubMed Abstract | Crossref Full Text | Google Scholar
17. Morello W, Proverbio E, Puccio G, Montini G. A systematic review and meta-analysis of the rate and risk factors for post-transplant disease recurrence in children with steroid resistant nephrotic syndrome. Kidney Int Rep. (2023) 8:254–64. doi: 10.1016/j.ekir.2022.10.030
PubMed Abstract | Crossref Full Text | Google Scholar
18. Liang R, Chen X, Zhang Y, Law C-F, Yu S, Jiao J, et al. Clinical features and gene variation analysis of COQ8B nephropathy: report of seven cases. Front Pediatr. (2022) 10:1030191. doi: 10.3389/fped.2022.1030191
留言 (0)