
記住我
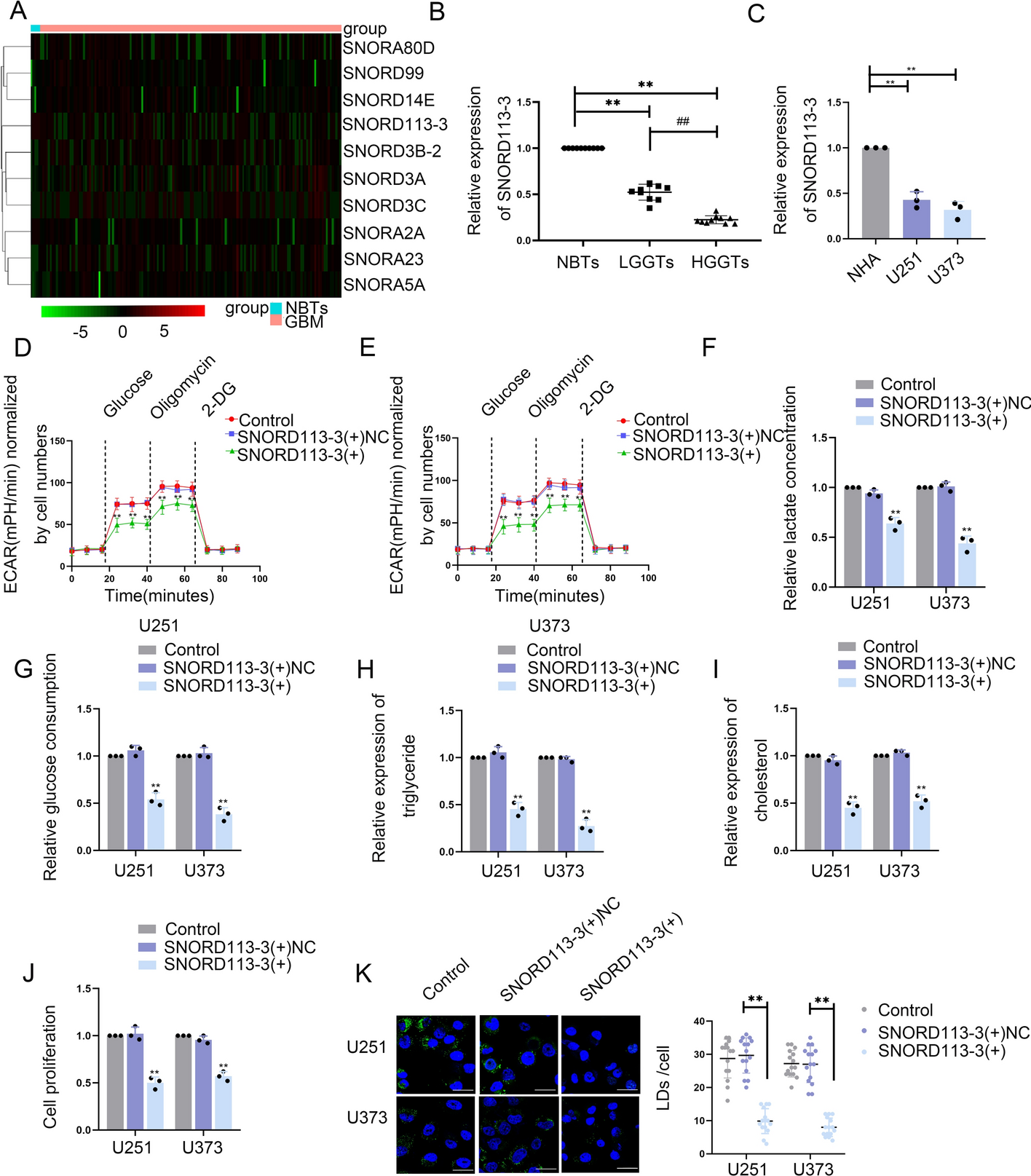


The TCGA database (https//:portal.gdc.cancer.gov/) was utilized to discern differentially expressed snoRNAs in comparison between GBM and normal brain tissues (NBTs) (Fig. 1A, Supplementary Table S2). To investigate the impact of these snoRNAs on glucose uptake and lactate production, they were knocked down in U373 and U251 cells. Among them, SNORD113-3 was chosen for further examination (Fig. S1A, B). qRT-PCR revealed that, compared with NHA, GBM cells, such as U87, U251, U373, and A172, presented decreased SNORD113-3 expression. U251 and U373 cell lines were selected for subsequent experiments (Supplementary Fig. S1C). The qRT-PCR analysis has demonstrated a significant reduction in the expression level of SNORD113-3 in GBM tissues, in comparison with its expression in NBT tissues. Additionally, a consistent decrease in SNORD113-3 expression was observed in correlation with the escalating pathological grade of glioma (Fig. 1B). Compared with NHAs, the expression of SNORD113-3 was notably downregulated in both U251 and U373 cells (Fig. 1C). Furthermore, confocal microscopy provided evidence of the nuclear localization of SNORD113-3 in U251 and U373 cells (Supplementary Fig. S1D).
Fig. 1
Screening for SNORD113-3, the expression of SNORD113-3, and its effects on glycolipid metabolism in GBM. A Heatmap with hierarchical cluster analysis of differentially expressed snoRNAs between non-tumor brain tissues (NBTs) and glioblastoma (GBM) tissues. P < 0.05, |log2FC|> 1. B SNORD113-3 expression was downregulated in tissues (NBTs [n = 10], LGGTs [n = 10], and HGGTs [n = 10]) via qRT-PCR. **P < 0.01 versus NBTs group; ##P < 0.01 versus LGGTs group. C SNORD113-3 expression level was analyzed in NHA cell, U251, and U373 cells via qRT-PCR. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus the NHA group. D, E The extracellular acidification rate (ECAR) was measured to demonstrate the effects of SNORD113-3 on glycolysis in U251 and U373 cells, and the glycolysis was calculated. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus SNORD113-3( +)NC group. F, G Lactate production and glucose uptake were measured in U251 and U373 cells after SNORD113-3 overexpression. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus SNORD113-3( +)NC group. H, I Intracellular triglyceride and cholesterol expression levels were measured after SNORD113-3 overexpression. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus SNORD113-3( +)NC group. J The effect of SNORD113-3 on proliferation was analyzed via CCK-8 assay. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus SNORD113-3( +)NC group. K Representative confocal fluorescence imaging of lipid droplets (LDs) stained by BODIPY 493/503 (green) in U251 and U373 cells. The nucleus (blue) was stained by DAPI. Scale bars = 10 µm. Data presented as mean ± SD (n = 15, each group). **P < 0.01 versus SNORD113-3( +)NC group. One-way ANOVA was used for statistical analysis
An investigation was conducted into the impact of SNORD113-3 on glycolipid metabolism and the proliferation of GBM cells. First, U251 and U373 cell lines with stable SNORD113-3 overexpression were constructed, and the group with the highest transfection efficiency was selected for further study (Supplementary Fig. S1E). Western blot was then conducted to measure the PKM2 and ACLY levels at the translational level in SNORD113-3-overexpression cells. PKM2 and ACLY expression was significantly reduced in SNORD113-3-overexpression cells (Supplementary Fig. S1F). We then measured the extracellular acidification rate, lactate, glucose, triglyceride, and cholesterol levels, and cell proliferation capacity of SNORD113-3-overexpression cells. In SNORD113-3-overexpression U251 and U373 cells, the aerobic glycolysis, lactate production, glucose utilization rate, lipid metabolism, and proliferation capacity were significantly reduced (Fig. 1D–J). Furthermore, fluorescence staining using BODIPY493/503 showed significantly reduced lipid droplets in U251 and U373 cells after SNORD113-3 overexpression (Fig. 1K).
ADAR2 is downregulated in GBM tissues and cells and suppresses glycolipid metabolism and GBM cell proliferationSNORD113-3 overexpression in U251 and U373 cells was associated with the upregulation of several genes as revealed by microarray analysis (Supplementary Fig. S2A). The expression level of mRNA for the RNA editing enzyme ADAR2 was increased significantly in U251 and U373 cells that overexpressed SNORD113-3, exhibiting a robust association with glucose uptake and lactate production. Consequently, ADAR2 was selected for further investigation (Supplementary Fig. S2B–E). Additionally, significant nuclear localization of ADAR2 in GBM cells was revealed by IF staining (Supplementary Fig. S2F). Western blots revealed that ADAR2 protein was more deficient in GBM tissues than in NBTs, and the decrease was more pronounced in higher pathological grades (Fig. 2A). Compared with NHAs, ADAR2 expression was downregulated significantly in both U251 and U373 cells (Fig. 2B). To further investigate the regulatory impact of ADAR2 on glycolipid metabolism in GBM cells, stable knockdown or overexpression of ADAR2 was successfully achieved in the U251 and U373 cell lines (transfection efficiency is shown in Supplementary Fig. S2G, H). ADAR2 overexpression resulted in dramatically reduced PKM2 and ACLY expression at the protein level (Supplementary Fig. S2I) and led to a notable reduction in the aerobic glycolysis, lactate production, glucose utilization, lipogenesis, and proliferation of GBM cells (Fig. 2C–J). Conversely, ADAR2 knockdown increased PKM2 and ACLY expression at the protein level significantly (Supplementary Fig. S2G) and promoted GBM cell growth and glycolipid metabolism (Fig. 2C–J).
Fig. 2
The expression of ADAR2 and its effects on glycolipid metabolism and proliferation in GBM. A ADAR2 protein levels were analyzed in NBTs, LGGTs, and HGGTs by western blot. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus NBTs group; ##P < 0.01 versus LGGTs group. B ADAR2 protein levels in NHA, U251, and U373 cells were detected via western blot. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus the NHA group. C, D The extracellular acidification rate (ECAR) was measured to demonstrate the effects of ADAR2 on glycolysis in U251 and U373 cells, and the glycolysis was calculated. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus ADAR2(–)NC group; ##P < 0.01 versus ADAR2( +)NC group. E, F Lactate production and glucose uptake were measured in U251 and U373 cells after ADAR2 knockdown or overexpression. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus ADAR2(–)NC group; ##P < 0.01 versus ADAR2( +)NC group. G, H Intracellular triglyceride and cholesterol expression levels were measured to evaluate the effect of ADAR2 on lipogenesis. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus ADAR2(–)NC group; ##P < 0.01 versus ADAR2( +)NC group. I The effect of ADAR2 on proliferation was analyzed via CCK-8 assay. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus ADAR2(–)NC group; ##P < 0.01 versus ADAR2( +)NC group. J Representative confocal fluorescence imaging of LDs stained by BODIPY 493/503 (green) in U251 and U373 cells after ADAR2 knockdown or overexpression. The nucleus (blue) was stained by DAPI. Scale bars = 10 µm. Data presented as mean ± SD (n = 15, each group). **P < 0.01 versus ADAR2(–)NC group; ##P < 0.01 versus ADAR2( +)NC group. Statistical analysis was performed using the one-way ANOVA method
SNORD113-3 promotes ADAR2 dimerization, glycolipid metabolism, and GBM cell proliferationWe conducted an investigation to delve into the association between SNORD113-3 and ADAR2. The bioinformatics database, starBasev2.0 (https://starbase.sysu.edu.cn/), indicates the existence of a potential binding interaction between ADAR2 and SNORD113-3. RIP showed SNORD113-3 enrichment in the ADAR2-immunoprecipitated samples compared with those immunoprecipitated using IgG (Fig. 3A). RNA pull-down experiments detected the binding of ADAR2 to SNORD113-3 in U251 cells (Fig. 3B). The data suggest that SNORD113-3 binds directly to ADAR2. While SNORD113-3 is indeed classified as a C/D box snoRNA, whose typical function involves 2′-O methylation within the nucleolus, our analysis utilizing NmSEER 2.0 prediction revealed no identifiable 2′-O methylation sites at either SNORD113-3 or ADAR2 binding locations (http://www.rnanut.net/nmseer-v2/). This suggests that SNORD113-3 may affect the nucleus through other molecular mechanisms independent of the rRNA modification induced by snoRNA.
Fig. 3
SNORD113-3 facilitated glycolipid metabolism of GBM cells by promoting ADAR2 dimerization. A An enrichment of SNORD113-3 in ADAR2 immunoprecipitated samples via RNA immunoprecipitation (RIP) assay. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus the anti-IgG group. B RNA pull-down assay followed by western blot showed the specific associations of ADAR2 with biotinylated-SNORD113-3 or antisense RNA. C Immunoprecipitation of FLAG-ADAR2 with HA-ADAR2 was performed using 293 T cells. D FLAG-ADAR2 and SNORD113-3 were coprecipitated with HA-ADAR2 in 293 T cells. E FLAG-ADAR2, HA-ADAR2, and SNORD113-3 were coprecipitated with anti-FLAG antibody, eluted with FLAG peptide, and then coprecipitated with the anti-HA antibody (n = 3, each group). F, G SNORD113-3 promote the binding of FLAG-ADAR2 to HA-ADAR2 both in vivo and in vitro (n = 3, each group). H The effects of ADAR2 stability were detected by cycloheximide (CHX) chase assays. Data presented as mean ± SD (n = 3, each group). **P < 0.01 versus SNORD113-3( +)NC group. I Representative confocal fluorescence imaging of LDs stained by BODIPY 493/503 (green) in U251 and U373 cells. The nucleus (blue) was stained by DAPI. Scale bars = 10 µm. Data presented as mean ± SD (n = 15, each group). **P < 0.01 versus control group; ##P < 0.01 versus SNORD113-3( +) + ADAR2(–)NC group; &&P < 0.01 versus SNORD113-3( +) + ADAR2( +)NC group. Statistical analysis was performed using the one-way ANOVA method
The dsRBDs of ADAR2 can directly bind to and edit dsRNAs; however, no editing sites for SNORD113-3 were identified in the RNA editing databases DARNED (http://darned.ucc.ie/about/) and REDIportal (http://srv00.recas.ba.infn.it/atlas/), indicating that SNORD113-3 is not a target gene for ADAR2 editing. As ADAR2 RNA editing depends on homodimerization, we wondered whether SNORD113-3 affected ADAR2 expression via ADAR2 homodimerization. The fundamental assumption underlying this conjecture is the capacity of SNORD113-3 to establish an association with an ADAR2 dimer. We first found that Flag-ADAR2 was coprecipitated with HA-ADAR2 in 293 T cells by co-IP assays (Fig. 3C). The simultaneous overexpression of FLAG-ADAR2, HA-ADAR2, and SNORD113-3 in 293 T cells followed by HA-ADAR2 immunoprecipitation assay showed significant enrichment of both FLAG-ADAR2 and SNORD113-3 (Fig. 3D). Furthermore, a rigorous two-step RIP analysis revealed that, during the first phase, the application of anti-FLAG antibody effectively coprecipitated HA-ADAR2 and SNORD113-3. Subsequently, the use of anti-HA antibody resulted in the concurrent precipitation of HA-ADAR2, FLAG-ADAR2, and SNORD113-3 (Fig. 3E). These experiments demonstrate that the dimer of SNORD113-3 and ADAR2 form a functional structure.
The authors further explored whether SNORD113-3 affects the dimerization of ADAR2. The results indicated that SNORD113-3 overexpression resulted in elevated relative expression of HA-ADAR 2 coupled to FLAG-ADAR 2 in either U251 cells or 293 T cells (Fig. 3F, G), indicating that SNORD113-3 promotes ADAR2 dimerization. Furthermore, upon examination of the results obtained from the cycloheximide chase assay, it was observed that the half-life of ADAR2 was significantly extended following the overexpression of SNORD113-3 (Fig. 3H), further confirming that SNORD113-3 is able to increase ADAR2 stability by promoting the formation of ADAR2 dimers and subsequently increasing ADAR2 protein expression.
The potential involvement of ADAR2 in the effect of SNORD113-3 expression on GBM cell glycolipid metabolism and proliferation was also investigated. After transfection of ADAR2(–), ADAR2( +), or the NC plasmids into stable SNORD113-3-overexpression cells, the ADAR2-overexpressed cells showed significantly reduced PKM2 and ACLY expression, glycolipid metabolism, and GBM cell proliferation (Supplementary Figs. 3I and S3A–H). However, the knockdown of ADAR2 reversed the suppressive effect exerted by SNORD113-3 on the expression of PKM2 and ACLY, resulting in the augmentation of glycolipid metabolism and the enhancement of GBM cell proliferation (Supplementary Figs. 3I and S3A–H).
PHKA2 upregulation in GBM tissues and cells and PHKA2 knockdown inhibits glycolipid metabolism and GBM cell proliferationMicroarray analysis revealed several significantly downregulated genes in ADAR2-overexpressed U251 and U373 cells (Supplementary Fig. S4A). The expression level of PHKA2 was decreased notably in U251 and U373 cells, and its downregulation was closely associated with glucose uptake and lactate production (Supplementary Fig. S4B–E). The RNA editing databases DARNED (http://darned.ucc.ie/about/) and REDIportal (http://srv00.recas.ba.infn.it/atlas/) have demonstrated that the process of A-to-I RNA editing can potentially take place within the 3′-UTR region of PHKA2 mRNA. Therefore, PHKA2 was selected for further study.
IF staining showed the PHKA2 cytoplasm localization in GBM cells (Supplementary Fig. S4F). Compared with NBTs, glioma tissues showed significantly higher PHKA2 expression, positively correlating with pathological grades (Fig. 4A). Consistent with the pattern observed in GBM tissues, GBM cells, including U251 and U373, demonstrated notably elevated PHKA2 expression levels (Fig. 4B). To elucidate the role of PHKA2 in glycolipid metabolism and GBM cell proliferation, we have established U251 and U373 cell lines in which PHKA2 has been knocked down or overexpressed. The levels of PHKA2 mRNA and protein expression within the cell lines are presented in Supplementary Fig. S4G, H. PKM2 and ACLY expression was reduced significantly in PHKA2-knockdown cells and suppressed glycolipid metabolism and GBM cell growth dramatically (Supplementary Fig. S4I; Fig. 4C–J). Conversely, PHKA2 overexpression enhanced PKM2 and ACLY expression, glycolipid metabolism, and GBM cell proliferation (Supplementary Fig. S4I; Fig. 4C–J).
Fig. 4
The expression of PHKA2 and its effects on glycolipid metabolism in GBM. A PHKA2 protein levels were analyzed in NBTs, LGGTs, and HGGTs by western blot. Data presented as mean ± SD (n = 3, each group), *P < 0.05 versus NBTs group; **P < 0.01 versus NBTs group; ##P < 0.01 versus LGGTs group. B PHKA2 protein levels in NHA, U251, and U373 cells were detected via western blot. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus the NHA group. C, D The extracellular acidification rate (ECAR) was measured to demonstrate the effects of PHKA2 on glycolysis in U251 and U373 cells, and the glycolysis was calculated. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus PHKA2(–)NC group; ##P < 0.01 versus PHKA2( +)NC group. E, F Lactate production and glucose uptake were measured in U251 and U373 cells after PHKA2 knockdown or overexpression. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus PHKA2(–)NC group; ##P < 0.01 versus PHKA2( +)NC group. G, H Intracellular triglyceride and cholesterol expression levels were measured to evaluate the effect of PHKA2 on lipogenesis. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus PHKA2(–)NC group; ##P < 0.01 versus PHKA2( +)NC group. I The effect of PHKA2 on proliferation was analyzed via CCK-8 assay. **P < 0.01 versus PHKA2(–)NC group; ##P < 0.01 versus PHKA2( +)NC group. Data presented as mean ± SD (n = 3, each group). J Representative confocal fluorescence imaging of lipid droplets (LDs) stained by BODIPY 493/503 (green) in U251 and U373 cells. The nucleus (blue) was stained by DAPI. Scale bars = 10 µm. Data presented as mean ± SD (n = 15, each group). **P < 0.01 versus PHKA2(–)NC group; ##P < 0.01 versus PHKA2( +)NC group. Statistical analysis was performed using the one-way ANOVA method
ADAR2 regulates PHKA2 expression by mediating A-to-I RNA editing of PHKA2 mRNAThe mechanism by which ADAR2 regulates PHKA2 expression was also investigated. RIP assays showed that PHKA2 mRNA can bind to ADAR2 (Fig. 5A). Editing sites for PHKA2 were identified in the RNA editing databases DARNED and REDIportal, suggesting PHKA2 as a potential A-to-I editing target gene. Given that A-to-I RNA editing events can potentially be facilitated by either ADAR2 or ADAR1, it is imperative that we eliminate the potential influence of ADAR1 on A-to-I RNA editing processes. The GBM-specific A-to-I RNA editing events that occurred in the 3′-UTR of PHKA2 mRNA were identified using Sanger sequencing. In the current study, no RNA editing changes were detected in U251 cells treated with ADAR2(−) NC, ADAR1(−), or ADAR1(−) NC, while a notable reduction in RNA editing was observed in U251 cells upon suppressing ADAR2 expression (Fig. 5B). The findings suggest that ADAR2 serves as the pivotal enzyme responsible for the specific A-to-I editing in the 3′-UTR region of PHKA2 mRNA, thereby playing a crucial role in GBM. Sanger sequencing analysis demonstrated that, upon knockdown of ADAR2, the A-to-I editing process within the 3′-UTR region of PHKA2 mRNA disappeared. Conversely, upon overexpression of ADAR2, the rate of A-to-I editing within the 3′-UTR region of PHKA2 mRNA was observed to be increased (Fig. 5C). The western blot assays demonstrated that the protein expression level of PHKA2 exhibited an elevation in response to the knockdown of ADAR2 expression, whereas it exhibited a decline upon the overexpression of ADAR2 (Fig. 5D). Given the possibility of A-to-I editing being implicated in the degradation of mRNA within the 3′-UTR region, a thorough examination of the half-life of PHKA2 mRNA was conducted. The results of the study demonstrate that overexpression of ADAR2 significantly reduced the half-life of PHKA2 mRNA in both U251 and U373 cells, thereby suggesting a critical regulatory role in this biological process (Fig. 5E). The above results demonstrate that ADAR2 could combine with PHKA2 and reduce PHKA2 mRNA stability through A-to-I editing, thereby reducing PHKA2 protein expression.
Fig. 5
PHKA2 facilitated glycolipid metabolism of GBM cells by ADAR2-induced A-to-I RNA editing. A An enrichment of PHKA2 mRNA in ADAR2 immunoprecipitated samples via RNA immunoprecipitation (RIP) assay. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus the anti-IgG group. B Sequence chromatograms of PHKA2 transcripts in U251 transduced with ADAR2(–)NC, ADAR2(–), or ADAR1(-)NC, ADAR1(–). Arrowheads indicate edited positions. Percentages indicate the calculated frequency of editing at selected positions. C Sequence chromatograms of the PHKA2 mRNA. Arrowheads indicate edited positions. D Western blot of ADAR2 and PHKA2 in U251 and U373 cells. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus ADAR2(–)NC group;##P < 0.01 versus ADAR2( +)NC group. E The half-life of PHKA2 mRNA at different times treated by ActD with ADAR2 overexpression in U251 and U373 cells. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus the ADAR2( +) group. F ECAR was used to measure the glycolysis and glycolytic capacity of U251 and U373 cells. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus control group; ##P < 0.01 versus ADAR2( +) + PHKA2(-)NC group; &&P < 0.01 versus ADAR2( +) + PHKA2( +)NC group. Statistical analysis was measured using the one-way ANOVA method.
To further investigate the functional significance of PHKA2, we knocked down or overexpressed PHKA2 in ADAR2-overexpressed U251 and U373 cells. We observed that PHKA2 knockdown enhanced the inhibition of ADAR2 overexpressed on PKM2 and ACLY protein expression (Supplementary Fig. S5A), while aerobic glycolysis, lactate production, glucose utilization, lipogenesis, and cell proliferation were reduced (Fig. 5F, Supplementary Fig. S5B–G). However, it was observed that the overexpression of PHKA2 reversed the inhibitory impact of ADAR2 overexpression on the expression of PKM2 and ACLY, and glycolipid metabolism and GBM cell proliferation were significantly augmented (Fig. 5F, Supplementary Fig. S5A–G).
EBF1 downregulation in GBM tissues and cells and EBF1 overexpression could suppress glycolipid metabolism and GBM cell proliferationTranscription factors associated with PKM2 and ACLY were analyzed using Cistrome (https://www.dbtoolkit.cistrome.org). PKM2, which is encoded by PKM genes, was identified as being linked to a total of 101 genes. Additionally, ACLY was discovered to be associated with 66 genes. Notably, 19 genes exhibited potential associations with both PKM2 and ACLY (Supplementary Fig. S6A; Supplementary File 1). The expression of these 19 genes after PHKA2 knockdown at the transcriptional and translational levels is shown in Supplementary Fig. S6B, C. EBF1 was significantly upregulated upon PHKA2 knockdown; therefore, EBF1 was selected for further investigation.
IF staining revealed that EBF1 is predominantly localized in the nuclei of GBM cells (Supplementary Fig. S6D). Compared with NBTs, the protein level of EBF1 in glioma tissues was observed to be relatively lower, with a further decline evident at higher pathological grades (Fig. 6A). In line with the glioma tissues, the GBM cells, such as U251 and U373, also exhibited significantly reduced EBF1 expression compared with NHAs (Fig. 6B). Subsequently, we conducted an analysis of the function of EBF1 in regulating glycolipid metabolism within GBM cells. Stable U251 and U373 cell lines with EBF1 knockdown or overexpression were developed. Supplementary Fig. S6E, F displays the EBF1 expression of transfected cells at transcriptional and translational levels. We observed significantly suppressed PKM2 and ACLY expression at the translational level (Supplementary Fig. S6G) and dramatically impaired glycolipid metabolism and markedly inhibited U251 and U373 cell growth (Fig. 6C–J). Conversely, increased PKM2 and ACLY expression, glycolipid metabolism, and GBM cell growth were observed after EBF1 knockdown (Supplementary Fig. S6G; Fig. 6C–J).
Fig. 6
The expression of EBF1 and its effects on glycolipid metabolism in GBM. A EBF1 protein levels were analyzed in NBTs, LGGTs, and HGGTs by western blot. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus the NBTs group; ##P < 0.01 versus the LGGs group. B EBF1 protein levels in NHA, U251, and U373 cells were detected via western blot. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus the NHA group. C, D The extracellular acidification rate (ECAR) was measured to demonstrate the effects of EBF1 on glycolysis in U251 and U373 cells, and the glycolysis was calculated. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus EBF1(–)NC group; ##P < 0.01 versus EBF1( +)NC group. E, F Lactate production and glucose uptake were measured in U251 and U373 cells after EBF1 knockdown or overexpression. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus EBF1(–)NC group; ##P < 0.01 versus EBF1( +)NC group. G, H Intracellular triglyceride and cholesterol expression levels were measured to evaluate the effect of EBF1 on lipogenesis. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus PHKA2(–)NC group; ##P < 0.01 versus PHKA2( +)NC group. I The effect of EBF1 on proliferation was analyzed via CCK-8 assay. **P < 0.01 versus EBF1(–)NC group; ##P < 0.01 versus EBF1( +)NC group. Data presented as the mean ± SD (n = 3, each group). J Representative confocal fluorescence imaging of lipid droplets (LDs) stained by BODIPY 493/503 (green) in U251 and U373 cells. The nucleus (blue) was stained by DAPI. Scale bars = 10 µm. Data presented as mean ± SD (n = 15, each group). **P < 0.01 versus EBF1(–)NC group; ##P < 0.01 versus EBF1( +)NC group. Statistical analysis was performed using the one-way ANOVA method
PHKA2 phosphorylated EBF1 on Y256 and reduced EBF1 stabilityThe possible interaction of PHKA2 with EBF1 was predicted using PPA-Pred2 (https://www.iitm.ac.in/bioinfo/PPA_Pred/) (Supplementary Fig. S7A). In U251 and U373 cells, IF staining revealed PHKA2 and EBF1 cytoplasm colocalization (Supplementary Fig. S7B). Additionally, co-IP assays showed an endogenous interaction of PHKA2 with EBF1 and FLAG-PHKA2 with GST-EBF1 in U251 and HEK-293 T cells, respectively (Fig. 7A, B). The PHKA2 and EBF1 direct binding was confirmed by an in vitro GST pull-down experiment using purified FLAG-PHKA2 and GST-EBF1 (Fig. 7C). Next, we knocked down or overexpressed EBF1 in PHKA2-deficient U251 and U373 cells. We found that EBF1 overexpression could enhance PKM2 and ACLY inhibition, glycolipid metabolism, and GBM cell proliferation in the PHKA2-knockdown group (Fig. 7D–F; Supplementary Fig. S7C–H), while EBF1 knockdown restored PKM2 and ACLY expression, increasing glycolipid metabolism and GBM cell proliferation (Fig. 7D–F; Supplementary Fig. S7C–H). Although EBF1 mRNA levels remained unchanged, the EBF1 protein was overexpressed following PHKA2 knockdown (Supplementary Fig. S7I, J). This observation indicates that the suppressive action of PHKA2 on EBF1 protein levels transpires following the translational process. A search using the phosphosite database (phosphosite.org/homeAction) identified multiple potential phosphorylation sites in EBF1 (Supplementary Fig. S7K). Together, these results indicate possible EBF1 phosphorylation by PHKA2. In vitro protein kinase assays confirmed that PHKA2 phosphorylates EBF1 (Fig. 7G). Using mass spectrometry analysis of the phosphorylated bands, the tyrosine at EBF1 position 256 (Y256) was identified as a specific PHKA2 phosphorylation site (Fig. 7H). Y256 mutation to alanine (Y256A) abolished the phosphorylated band (Fig. 7I). Subsequent in vivo experiments using customized specific phosphoantibodies showed that EBF1-Y256A phosphorylation was lower than that of the wild-type EBF1 (EBF1-WT), while EBF1-Y256E, a mutant in which Y256 is replaced by glutamate, had higher phosphorylation levels (Supplementary Fig. S7L). PHKA2 overexpression resulted in increased EBF1 phosphorylation at Y256, and this higher EBF1 phosphorylation level led to lower protein expression (Supplementary Fig. S7M, N). Furthermore, compared with EBF1-WT, EBF1-Y256A and EBF1-Y256E exhibited a significantly prolonged and shortened half-life, respectively (Fig. 7J).
Fig. 7
The effects of EBF1 phosphorylated by PHKA2 on glycolipid metabolism and proliferation in GBM. A Lysates of U251 cells were subjected to immunoprecipitation (IP) and immunoblotting (IB) with PHKA2 and EBF1 antibodies. B Lysates of 293 T cells transfected with FLAG-PHKA2 and GST-EBF1 plasmids were subjected to IP and IB with FLAG tag and GST tag antibodies. C The direct interaction between PHKA2 and EBF1 was confirmed by GST pull-down assays. GST protein functioned as a negative control. D, E ECAR was used to measure the glycolysis and glycolytic capacity of U251 and U373 cells. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus control group; ##P < 0.01 versus PHKA2(–) + ENF1( +)NC group; &&P < 0.01 versus PHKA2(–) + EBF1(–)NC group. F Representative confocal fluorescence imaging of LDs stained by BODIPY 493/503 (green) in U251 and U373 cells. The nucleus (blue) was stained by DAPI. Scale bars = 10 µm. Data presented as mean ± SD (n = 15, each group). **P < 0.01 versus control group; ##P < 0.01 versus PHKA2(–) + EBF1( −)NC group; &&P < 0.01 versus PHKA2(-) + EBF1( +)NC group. G Left panel, in vitro kinase assays were performed and detected by autoradiography (arrow, phosphorylated band). Right panel, proteins were visualized by Coomassie brilliant blue (CBB) staining. H The phosphorylated bands were subjected to mass spectrometry, and Y256 was identified. I In vitro kinase assays and CBB staining were conducted after Y256 mutation. J The effects of Y256 phosphorylation on EBF1 stability were detected by cycloheximide (CHX) chase assays. Data presented as mean ± SD (n = 3, each group). **P < 0.01 versus EBF1-WT group. Statistical analysis was performed using the one-way ANOVA method
EBF1 inhibited PKM2 and ACLY transcription through directly binding to their promotersThe underlying molecular mechanisms behind PKM2 and ACLY suppression by EBF1 were further examined. Using the UCSC Genome Browser (http://genome.ucsc.edu/), we analyzed the transcription start sites of ACLY and PKM2, and several potential binding sites within the first 1000 bp of the ACLY and PKM2 promoter regions for EBF1 were discovered. Meanwhile, direct EBF1 binding to the PKM2 and ACLY promoter region was also observed in vitro (Fig. 8A–D). Further experiments revealed that EBF1 inhibited PKM2 and ACLY transcriptional expression. These findings demonstrate that the transcriptional regulation of PKM2 and ACLY is mediated by EBF1, which serves as a critical regulator in governing the biological behavior exhibited by GBM cells.
Fig. 8
EBF1 directly bound to the promoter regions of PKM2, ACLY and transcriptionally suppressed their expression. A The putative EBF1 binding site is indicated in the PKM2 promoter region (above). Chromatin immunoprecipitation (ChIP) assay showed the products amplified putative EBF1-binding sites of PKM2 (below). B Schematic diagram of luciferase reporter construction and PKM2 relative luciferase activity measured in cells cotransfected with the PKM2 promoter (−1000 to 0 bp) (or PKM2 promoter-deleted putative EBF1 binding site) and pEX3 empty vector or pEX3-EBF1. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus pEX3 empty vector group. C The putative EBF1 binding site is indicated in the ACLY promoter region (above). ChIP assay showed the products amplified putative EBF1-binding sites of ACLY (below). D Schematic diagram of luciferase reporter construction and ACLY relative luciferase activity measured in cells cotransfected with the ACLY promoter (−1000 to 0 bp) (or ACLY promoter-deleted putative EBF1 binding site) and pEX3 empty vector or pEX3-EBF1. Data presented as mean ± SD (n = 3, each group), **P < 0.01 versus pEX3 empty vector group. One-way ANOVA was used for statistical analysis
Simultaneous SNORD113-3 and ADAR2 overexpression and PHKA2 knockdown suppressed tumor growth and prolonged nude mouse survivalTo further examine the effect of SNORD113-3, ADAR2, and PHKA2 on tumor progression in vivo, GBM-SNORD113-3( +) cells, ADAR2( +) cells, PHAK2(–) cells, or a combination of them was injected to establish a mouse xenograft model. Compared with the control group, SNORD113-3( +), ADAR2( +), and PHKA2(–) significantly reduced the volume, with the smallest sizes observed in mice injected with PHKA2(–) or a combination of SNORD113-3( +) and ADAR2( +) (Fig. 9A, B).
Fig. 9
Overexpression of SNORD113-3 and ADAR2 with knockdown of PHKA2 suppressed tumor growth and prolonged survival in nude mice. A Subcutaneously xenografted nude mice injected with different treated cells are shown (above). Representative tumors from each group are shown (below). B Tumor growth curves are shown. Tumor size was recorded every 5 days, and tumors were extracted at 45 days after injection. **P < 0.01 versus control group; ##P < 0.01 versus SNORD113-3(+) + ADAR2(+) + PHKA2(−) group by two-way ANOVA. C Survival curves of nude mice with orthotopic xenografts are shown. Data presented as mean ± SD (n = 8, each group). **P < 0.01 versus control group; ##P < 0.01, &&P < 0.01, ^^P < 0.01, SNORD113-3(+), ADAR2(+) or PHKA2(–) group compared with the SNORD113-3(+) + ADAR2(+) + PHKA2(–) group, respectively
Next, we conducted a thorough analysis of the survival rates of mice implanted with the specified GBM cells. Compared with the control group, mice injected with SNORD113-3(+), ADAR2(+), and PHKA2(–) showed significantly longer survival times. Notably, the mice injected with all three factors displayed the longest survival duration. (Fig. 9C). Figure 10 shows a schematic diagram of the molecular mechanism by which SNORD113-3 mediates ADAR2 A-to-I RNA editing of PHKA2 mRNA to promote EBF1-Y256 phosphorylation, regulating glycolipid metabolism and GBM cell growth.
Fig. 10
SNORD113-3 mediates ADAR2 A-to-I editing of PHKA2 mRNA, promoting EBF1-Y256 phosphorylation in the regulation of glycolipid metabolism and proliferation of GBM cells: schematic diagram
留言 (0)