
記住我
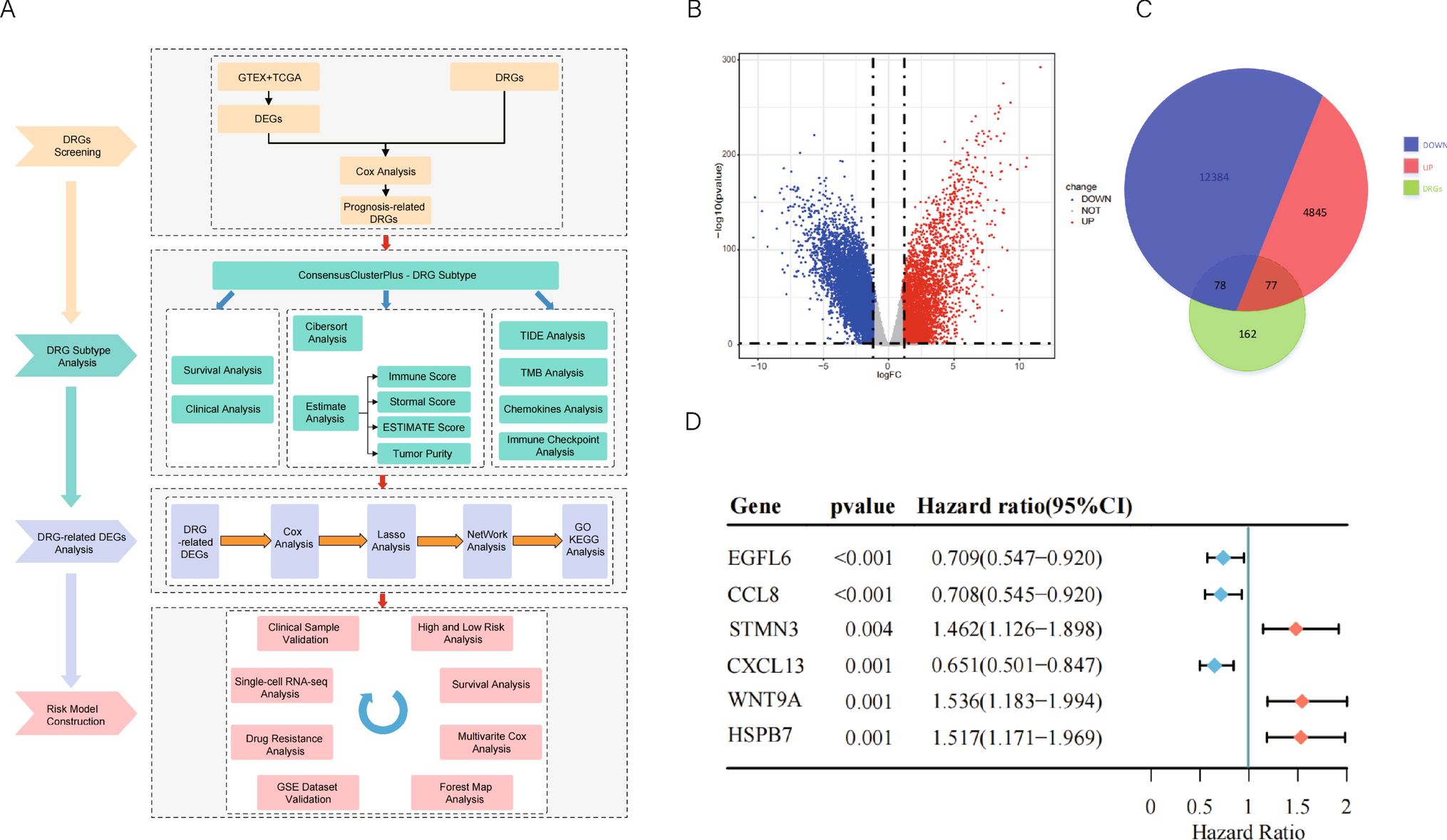




The study's workflow is shown in Fig. 1A. Transcriptome data from 427 OC patients and their clinical characteristics were retrieved from the TCGA-OV database, with transcriptome data from 88 normal tissues obtained from the GTEx database as controls. LIMMA differential expression analysis identified 17,384 differentially expressed genes (DEGs), including 4922 upregulated and 12,462 downregulated genes (Fig. 1B). We then obtained 317 disulfidptosis-related genes (DRGs) from the GeneCards database and recent literature. Intersection analysis with the DEGs revealed 155 DRGs in both normal and OC tissues, with 77 upregulated and 78 downregulated (Fig. 1C). Univariate Cox regression analysis (P < 0.01) identified six genes—CCL8, CXCL13, EGFL6, HSPB7, STMN3, and WNT9A—as having prognostic predictive value.
Fig. 1
The workflow of the experiment and identification of differentially expressed DRGs. A Workflow of the experiment. B Volcano Plot visualization for differential genes of normal tissue (GTEX) and cancer tissue (TCGA-OV) by employing |logFC|> 1 and FDR < 0.05 parameters. C Venn diagram of DRGs identification. D Six DRGs (CCL8, CXCL13, EGFL6, HSPB7, STMN3, WNT9A) with prognostic predictive values were found utilizing univariate regression analysis
3.2 Classification of molecular subtypes according to DRGsBased on the six prognostically relevant DRGs, the cancer patient cohort (n = 427) was subjected to consensus clustering. The optimal rank, determined by the covariate correlation coefficient, was k = 2 (Fig. 2A, B). With k = 2, two subtypes were identified. PCA and t-SNE validated the classification into these two distinct subtypes (Fig. 2C, Fig. S1A). Thus, the TCGA-OV samples were classified into Cluster 1 (n = 224) and Cluster 2 (n = 151).
Fig. 2
Classification of molecular subtypes according to DRGs; the assessment and validation of subtype characterization. A, B Unsupervised consensus cluster analysis identified the best DRG subtypes in ovarian cancer as k = 2. C PCA analysis assesses the effectiveness of clustering. Comparison between two subtypes in (D) DRGs expression level E Kaplan–Meier analysis of survival prognosis of two DRG subtypes F Clinical features G Tumor mutational burden and tumor purity H Estimate scores, immune score and stromal score (*P < 0.05; **P < 0.01; ***P < 0.001)
3.3 Clinical assessment and validation of subtype characterizationThe six prognostic DRGs identified were compared between the two subtypes (Fig. 2D). Cluster1 exhibited higher expression levels of CCL8, CXCL13, and EGFL6, while Cluster2 showed increased levels of HSPB7, STMN3, and WNT9A. Overall survival analysis revealed that Cluster2 had worse overall survival compared to Cluster1 (Fig. 2E). A clinical assessment of age, histologic grade, tumor status, and clinical stage for both subgroups is shown in Fig. 2F. Tumor mutational burden (TMB), analyzed using TCGA-OV data, was lower in Cluster2, suggesting better immunotherapy responsiveness and prognosis for Cluster1 (Fig. 2G). Additionally, the ESTIMATE algorithm was used to assess stromal and immune cell infiltration, as well as tumor purity. Cluster2 had lower stromal, immune, and estimated scores, along with higher tumor purity, indicating a poorer prognosis (Fig. 2G, H), further supporting the clinical assessment.
3.4 Immune assessment between the two DRG subtypesTo explore the cell infiltration landscape of the tumor immune microenvironment, the CIBERSORT algorithm was used to determine the composition of 22 immune cell types in the two subtypes. The results revealed an increased presence of CD8 + T cells, activated CD4 + memory T cells, follicular helper T cells, γδT cells, and M1 macrophages, alongside a reduction in M0 macrophages in Cluster 1 compared to Cluster 2 (Fig. 3A). The immune infiltration algorithm TIMER (Fig. S1B) and xCell (Fig. S1C) also obtained similar results. Immune checkpoints (ICBs) are immunosuppressive molecules expressed on immune cells that modulate immune activation and serve as biomarkers for immunotherapy. HAVCR2, VTCN1, BTLA, CD274, CTLA4, LAG3, LILRB1, PDCD1, SIRPA, and TIGIT are key immune checkpoint genes. Except for SIRPA, which showed no significant difference, the other genes exhibited reduced expression in Cluster2 (Fig. 3B), suggesting Cluster2 represents an immune-cold subtype with diminished susceptibility to immunotherapy and a poorer prognosis. Analysis of chemokine expression profiles from the TCGA database revealed decreased levels of CCR1, CCR2, CCR4, CCR5, CCR7, CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL8, CXCL9, CXCL10, CXCL11, CXCL13, CXCR2, and CXCR4 in Cluster2, with no significant difference in CXCL14 (Fig. 3C). This collective downregulation implies a worse prognosis for Cluster2 patients. Additionally, we used the Tumor Immune Dysfunction and Rejection (TIDE) algorithm to predict ICB efficacy and the likelihood of tumor immune escape. Cluster2 had a higher TIDE score, indicating reduced ICB efficacy and a higher likelihood of immune escape, further suggesting a poorer prognosis (Fig. 3D).
Fig. 3
Immunological analysis between the DRG subtypes. Comparison about A tumor-infiltrating cells B immune checkpoint genes C chemokines D TIDE score. (P < 0.05 was considered to be significant) (*P < 0.05; **P < 0.01; ***P < 0.001)
3.5 Construction of the risk score modelLIMMA differential expression analysis of the two subtypes identified 428 DEGs, with 378 upregulated in Cluster1 and 50 in Cluster2 (Fig. 4A). After Lasso-Cox regression analysis (Fig. 4B, C), 15 prognosis-related genes were identified, and six hub genes were determined using Network Analyst: CD38, IL21, CD40LG, IFNB1, IL1B, and IGF2 (Fig. 4E). The expression profiles of the 15 genes are shown in the heatmap (Fig. 4D), where CD38, IL21, CD40LG, IFNB1, and IL1B were poorly expressed in Cluster2, and IGF2 was highly expressed. KEGG and GO enrichment analyses indicated that these six hub genes are mainly involved in immune cell metabolism and cytokine regulation (Fig. 4F, G). Based on these genes, a risk score model was created, and the risk value of each sample was calculated. Risk score = −0.4029086 × IFNB1 + 0.2958303 × IGF2 + (−0.3709597) × CD40LG + 0.2803076 × IL1B × (−0.3618734) × IL21 + (−0.5388026) × CD38. All OC patients were classified into high-risk and low-risk groups using the median risk cutoff.
Fig. 4
Differential expression between DRG subtypes and identification of signature gene sets. A volcano plot by employing |logFC|> 1 and FDR < 0.05 parameters; Lasso-Cox regression analysis B, C LASSO regression analysis of DEGs which are most significantly correlated with OS; D heat map display of DEGs in DRG subtypes; E network analysis of the prognostic and hub genes identification; GO (F) and KEGG (G) analysis about DRG-related DEGs
3.6 Validation of the risk score model based on signature genesThe expression profiles of the six signature genes in both groups were consistent with those from a previous study (Fig. 5A). A subset of 355 patients with complete data on age, stage, and grade was extracted from the TCGA-OV dataset. Multivariate Cox regression analysis, including age, grade, and stage as covariates, confirmed that the risk score model derived from the six signature genes was an independent prognostic factor in the TCGA-OV cohort (HR = 1.13, 95% CI 1.074–1.20, P < 0.001) (Fig. 5B). An increase in the risk score was associated with higher patient mortality (Fig. 5C). Kaplan–Meier survival analysis showed significantly better survival in the low-risk group compared to the high-risk group, and the ROC curve supported the model’s predictive value for 1-year, 3-year, and 5-year overall survival (OS) (Fig. 5D). The Sankey diagram illustrated the correlation between the risk score and clinical parameters (age, grade, stage, tumor status), confirming the worse prognosis of the high-risk group (Fig. 5E). Using the Genomics of Cancer Drug Sensitivity (GDSC) dataset, we compared the half-maximal inhibitory concentrations (IC50) of eight classic OV chemotherapy agents. The high-risk group showed increased resistance to all agents, indicating reduced chemotherapy efficacy and a poorer prognosis (Fig. 5F). Additionally, we analyzed the relationship between immune cell infiltration and risk scores. Mast cells and M0 macrophages were positively correlated with the risk score, while other immune cell types, such as CD8 + T cells, CD4 + T cells, and Tfh cells, were negatively correlated (Fig. 5G).
Fig. 5
Validation of the risk score model based on signature genes. A Rank score and survival status observation between high and low-risk groups. B Multivariate Cox regression analysis of this risk score model together with covariates. C Kaplan–Meier OS curves were created for high and low-risk groups. D The analysis's False Positive Rate (AUC) was negligible, which is a strong indication of how well the risk score performed in terms of predicting OS for 1 year, 3 years, and 5 years. E The Sankey diagram visualizes the correlation between the risk score and clinical parameters. F Assessment of chemotherapy sensitivity between the high and low risk-groups. G Correlations between risk score and immune cell types (*P < 0.05; **P < 0.01; ***P < 0.001)
3.7 Assessment and validation of the risk score modelTo validate the efficacy of the proposed score model, two independent datasets, GSE13876 and GSE26712, comprising 415 and 153 OC samples respectively, were extracted from the GEO database. Both datasets were categorized into high-risk and low-risk groups based on the median risk cutoff value. Survival analyses consistently showed that the high-risk group had significantly poorer survival compared to the low-risk group (Fig. 6A, C). Additionally, the area under the curve (AUC) values for 1-year, 3-year, and 5-year overall survival (OS) in the ROC curves of both datasets exceeded 50% (Fig. 6B, D), confirming the model's strong predictive ability in independent cohorts.
Fig. 6
Validation of the risk model using external dataset by observing A, C overall survival and B, D roc curves between two risk groups. All validation cohort data comes from GEO data sets GSE13876 and GSE26712. (P value was calculated to define the significance on the analysis)
3.8 Comparison of signature gene expression in conjunction with single-cell sequencingBy integrating single-cell analysis data from multiple GEO datasets, we conducted a comparative assessment of the six signature genes across distinct cellular subpopulations. The findings showed that in the tumor microenvironment (TME) (Fig. 7A, B). IGF2 was predominantly expressed in malignant cells and stromal cells, especially malignant cells. Notably, CD38, CD40LG and IL1B were significantly expressed in both stromal and immune cells, while IL21 and IFNB1 exhibited limited expression. The predominant expression of IGF2 in malignant cells, and CD38, CD40LG, and IL1B in nonmalignant cells, suggests a potential role in regulating tumor development, consistent with the gene expression patterns observed in the high- and low-risk groups.
Fig. 7
Utilizing the UMAP method, the distribution of cells in tumor microenvironment was visualized, The mRNA expression of GEO datasets A GSE154600 and B GSE130000
3.9 The risk score corresponds with disease progression-free survival and platinum resistanceTo transition from bioinformatics predictions to practical insights, we experimentally validated the expression of key genes in clinical samples to confirm their biological relevance and association with clinical outcomes, thereby strengthening our prognostic model. Using qPCR and IHC, we assessed the expression of CD38, CD40LG, IGF2, IL1B, IL21, and IFNB1, evaluating their correlation with clinical variables and the risk score. Additionally, IHC was used to evaluate K-i67 expression in tumor tissues, providing an assessment of cellular proliferative capacity.
Our findings indicated that patients with epithelial OC showed significantly higher IGF2 and IL1B expression compared to those with borderline tumors, while CD38, CD40LG, IL21, and IFNB1 showed no significant differences (Fig. 8A). In late-stage OC tissues, CD38, CD40LG, IL21, and IFN1 levels were higher compared to early-stage tissues, whereas IL1B remained unchanged. IGF2 was notably higher in malignant tissues, particularly in early stages (Fig. 8B). Based on the prognostic model, the OC group was divided into high and low-risk groups using the median risk score. The low-risk group had longer progression-free survival than the high-risk group (P = 0.058) (Fig. 8C). Patients were also classified based on the platinum-free interval: platinum-resistant (< 6 months) or platinum-sensitive (> 6 months). The platinum-sensitive group had significantly lower risk scores than the platinum-resistant group (P < 0.05) (Fig. 8D). Figure 8E illustrates representative images depicting high, medium, and low expression levels of CD38, CD40LG, IGF2, IL1B, IL21, IFNB1, and Ki-67. The expression levels of each hub gene at the protein level were compared between the high- and low-risk groups (Fig. 8F). It was found that CD38 expression was significantly higher in the low-risk group compared to the high-risk group (P < 0.01), whereas IGF2 and IL1B exhibited higher expression in the high-risk group (P < 0.01). No significant differences were observed for IL21, CD40LG, and IFNB1 between the two groups. Additionally, the high-risk group showed elevated Ki-67 expression (P < 0.05), indicating increased proliferative activity (Fig. 8G).
Fig. 8
The risk score corresponds with disease progression-free survival and platinum resistance. The expression of characteristic genes differs between cancerous and noncancerous tissues (A), early-stage and advanced ovarian cancer (B). C Kaplan–Meier analysis of progression-free survival rate of low-risk group and high-risk group. D The comparison of risk score between platinum-resistant group and platinum-sensitive group. E Representative IHC images: Immunohistochemical staining depicting high, medium, and low expression levels of CD38, CD40L, IGF2, IL1B, IL21, IFNβ, and Ki-67 in tumor tissues. F Comparison of protein expression levels of the hub genes between high- and low-risk groups. G Ki-67 protein expression in high- and low-risk groups. (*P < 0.05; **P < 0.01; ***P < 0.001)
留言 (0)