
記住我
The complex nature of cancer biology imposes a major challenge in cancer research and, consequently, the development of effective treatment regimes. The World Health Organization (WHO) estimated that 10 million patients globally died from various forms of cancer in 2020 alone. Many clinical trials do not provide significant success despite significant advances in diagnosis and innovative therapy methods (Karimi et al., 2022). Even though targeted therapy has been a successful approach in treating cancer, heterogeneous cancer still has a variety of clinical profiles and molecular alterations. Certain genetic alterations in cancer targets can make drugs more effective or, more often, cause them to become resistant to treatment (Jin et al., 2019). Drug resistance caused by mutations is a common occurrence in cancer. Thus, the mutation profile of patient malignancies plays a major role in determining the effectiveness of targeted therapy. Accurate molecular and genetic profiling of tumour cells is becoming a crucial step before implementing targeted therapy in patients (Jin et al., 2019).
The oncoprotein B-cell lymphoma-2 (Bcl-2) family proteins control apoptosis and are implicated in various tumour progressions (Rosser et al., 2003; Goff et al., 2013). This gene was the first to promote prolonged cell survival and growth rather than boost proliferation, demonstrating the importance of inhibiting cell death in tumorigenesis (Cory and Adams, 2002). Bcl-2 inhibits cytochrome c (cyt-c) release from the mitochondria, preventing caspases involved in apoptosis from activating (Yin et al., 1994). Bcl-2 overexpression or aberrant expression has been associated with many cancers’ emergence, progression, and relapse (Delbridge et al., 2016; Kitada et al., 2002). Consequently, Bcl-2 activity and protein levels have emerged as essential measures for determining the success or failure of clinical treatment and predicting patient outcomes (Delbridge et al., 2016). The sensitivity of malignant tumor cells to apoptosis can be efficiently boosted by either lowering Bcl-2 protein levels or suppressing Bcl-2 function (Qian et al., 2022). Multidrug resistance (MDR) in cancer cells can be overcome by selectively inhibiting Bcl-2, resulting in cell cycle arrest, senescence, and eventual cell death in response to radiotherapy and chemotherapy (Tang et al., 2020; Wang et al., 2020). Therefore, inhibition of Bcl-2 inactivation has become a highly attractive strategy in the battle against cancer, and BH3 mimetics are the main category of promising therapeutic agents (Perini et al., 2018; Delbridge and Strasser, 2015). BH3 mimetics inhibit Bcl-2 activity by competing with its physiological ligands, BH3 domain-containing pro-apoptotic proteins, at the hydrophobic (binding) groove (Czabotar et al., 2014).
Despite the promising initial clinical effectiveness of BH3 mimetic agents in various cancers, the mutation is a common way cancer cells evade therapies (Roberts et al., 2016; Stilgenbauer et al., 2018). The most common mutation is a change from glycine to valine at amino acid position 101 (G101V), which substantially decreases Bcl-2 affinity towards the BH3 mimetics agent (Venetoclax) and prevents the drug from displacing pro-apoptotic mediators from Bcl-2 in the cells (Blombery et al., 2019; Blombery et al., 2020). Most human genetic variations are attributable to single nucleotide polymorphisms (SNPs) (Dakal et al., 2017). This genetic variation generated by SNPs in genetic codons influences the translation outcome, resulting in a mutant protein with a different structure and function. Nevertheless, not all SNPs impact protein function and structure; a few are harmful, but many are not (Kucukkal et al., 2015).
Bioinformatics offers enormous array of databases and techniques that are necessary for the analysis, integration, and interpretation of cancer multi-omics data (Jiménez-Santos et al., 2022). It is noteworthy that in silico techniques have recently emerged as valuable tool to assess the distinct genomic alterations and transcriptome profiles of tumors, as well as understanding the underlying mechanisms of cancer (Yalcin-Ozkat, 2021; Edelman et al., 2010; Elamin et al., 2024).
Herein, combined in silico, bioinformatic approaches and molecular dynamics simulations were employed to comprehensively analyze the genomic and proteomic changes in Bcl-2 (Figure 1) and their implications on carcinogenicity. In order to do cross-validation and ensure the validity of the generated data, we choose to employ a variety of bioinformatics algorithms for each type of analysis we carried out in this work. Several mutations have been analyzed for their potential impact on the genesis and progression of cancer, and their deleterious effects on Bcl-2 structure and function have been described. Subsequently, the most deleterious mutations, Bcl-2G101V and Bcl-2F104L, were selected for further dynamic analysis to probe their impact on the protein conformational landscape using molecular dynamics (MD) simulations and post-dynamic analyses.
Figure 1. Flowchart of the different types of analyses and approaches employed in this study.
We believe that the extensive and multifaceted analyses provided in this study will offer a thorough grasp of the effects of deleterious Bcl-2 gene mutations on the apoptotic machinery and their implications for carcinogenesis. This understanding will then inform future directions in drug design and the development of anti-cancer therapeutics.
2 Methods2.1 Generation of the datasetsThe Bcl-2 FASTA sequence was obtained from UniProt (UniProt ID: P10415) (https://www.uniprot.org/) (Bateman et al., 2017). The dbSNP (https://www.ncbi.nlm.nih.gov/snp/) and Ensembl (https://www.ensembl.org/) databases and an extensive literature search were used to compile the list of mutations (Sherry et al., 2001; Hubbard et al., 2002). Gene synonyms (Bcl-2, PPP1R50) (transcript ID: ENST00000333681.5) of the Bcl-2 protein were selected for this study. Duplicate variants and other redundant data were excluded from the analysis. High-resolution crystal structures of the Bcl-2 protein, both wild-type and mutated (G101V and F104L) (PDB ID:6O0K, 6O0L, and 6O0M), were obtained from the Protein Data Bank (https://www.rcsb.org/) (Birkinshaw et al., 2019).
2.2 Sequence-based analyses for point mutationWe utilised eight different bioinformatics tools to obtain a reliable cross-validated sequence-based analysis to determine the deleterious effects of residue mutations on the protein. These are, the Sorting Intolerant From Tolerant (SIFT) algorithm (https://sift.bii.a-star.edu.sg) which determines the deleterious effects of residue mutations on proteins (Kumar et al., 2009); Polymorphism Phenotyping 2 (PolyPhen-2) (http://genetics.bwh.harvard.edu/pph2/) (Adzhubei et al., 2013), which is tailored to the study of high-throughput Next-Generation Sequencing (NGS) data and features multiple sequence alignments and classifiers based on machine learning; Combined Annotation Dependent Depletion (CADD) (https://cadd.gs.washington.edu/) that is designed to estimate the deleterious effect of residue variation on protein sequences (Rentzsch et al., 2019); Rare Exome Variant Ensemble Learner (REVEL) (https://sites.google.com/site/revelgenomics/) (Ioannidis et al., 2016); MetaLR (https://sites.google.com/site/jpopgen/dbNSFP) which predicts the deleteriousness of missense variants using logistic regression, which incorporates nine independent variant deleteriousness scores and allele frequency information (Liu et al., 2016); Mutation Assessor (http://mutationassessor.org/r3/) uses the evolutionary conservation of the impacted residues in protein homologs to speculate on the functional consequences of residue changes in proteins (Reva et al., 2011); Functional Analysis Through Hidden Markov Models (FATHMM) which is a high-throughput web server capable of predicting the functional consequences of both coding variants, that is, non-synonymous single nucleotide variants (nsSNVs) and non-coding variants in the human genome (http://fathmm.biocompute.org.uk/); and Predict-SNP (https://loschmidt.chemi.muni.cz/predictsnp1/) (Bendl et al., 2014).
2.3 Structure-based analyses for point mutationVarious algorithms were employed to predict the effect of missense mutations on the protein stability. These include, mCSM (https://biosig.lab.uq.edu.au/mcsm/) which uses various residues atomic distance patterns to train the predictive models (Pires et al., 2014a); Site-directed mutator2 (SDM2) (http://marid.bioc.cam.ac.uk/sdm2) which can also estimate the relative stability of wild-type and mutated protein structures by comparing them to known homologous 3D structures; DUET (http://biosig.unimelb.edu.au/duet/) which uses Support Vector Machines (SVM) to produce a consensual estimate (Pires et al., 2014b); PremPS (https://lilab.jysw.suda.edu.cn/research/PremPS/) which estimates changes in the Gibbs free energy of protein unfolding to assess the impact of single mutations on protein stability (Chen et al., 2020); CUPSAT (http://cupsat.tu-bs.de/) (Parthiban et al., 2006); ENCoM (https://labworm.com/tool/encom) (Frappier et al., 2015); MutPred2 (http://mutpred.mutdb.org/) (Pejaver et al., 2020); and DynaMut (https://biosig.lab.uq.edu.au/dynamut/) which takes the changes in vibrational entropy into account (Rodrigues et al., 2018).
2.4 Disease phenotype prediction analysisSeveral machine learning and neural network algorithms were employed for disease phenotype prediction. These include, PhD-SNP (https://bio.tools/phd-snp) which uses neural networks that have been trained on a large library of standard and pathogenic mutations (Capriotti and Fariselli, 2017); Protein ANalysis THrough Evolutionary Relationships (PANTHER) (http://www.pantherdb.org/) which is designed to estimate the likelihood of a particular non-synonymous (residue changing) coding SNP that causes a functional impact on the protein (Thomas et al., 2022); SNPs and GO (https://snps.biofold.org/snps-and-go/) is another a precise technique that uses the associated protein functional annotation to determine whether or not a variation is associated with a disease based on a protein sequence (Capriotti et al., 2013); PMut (http://mmb.irbbarcelona.org/PMut/) which identifies pathogenic protein variants with up to 80% predictive accuracy in humans (López-Ferrando et al., 2017); and Meta-SNP (https://snps.biofold.org/meta-snp/) which is a randomised forest-based classification algorithm that distinguishes between polymorphic non-synonymous SNVs and disease-related one.
2.5 Post-transcriptional modification (PTM) sites predictionPTM site predictions comprised several rearranged residues that produced many proteins. Ubiquitination, phosphorylation, and methylation are some of the PTM sites that have been characterised. These sites are essential in vital cellular organising processes such as pathological signaling cascades and protein-protein interactions. Thus, PTM prediction assisted in elucidating whether genetic variants were associated with or contributed to disease pathogenesis. We used four tools for this purpose, namely,; NetPhos 3.1 (https://services.healthtech.dtu.dk/service.php?NetPhos-3.1); Group-based Prediction System (GPS) 6.0 (http://gps.biocuckoo.cn/) (Xue et al., 2005); BDM-PUB (http://bdmpub.biocuckoo.org/) which is for protein ubiquitination site prediction using the Bayesian Discriminant Method; and UbPred (http://www.ubpred.org/).
2.6 Gene-gene interaction network analysisThe gene function can be better understood by studying the genes with which it interacts. The GeneMANIA and STRING databases were used to investigate the relationship between the Bcl-2 gene and other genes and to predict the effect of Bcl-2 nsSNPs on other associated genes. GeneMANIA (https://genemania.org/) is a database for identifying genes related to input genes using an extensive set of functional association data (Warde-Farley et al., 2010). These association data included co-expression, colocalisation, pathways, protein domain similarity, and interactions between proteins and genes. GeneMANIA can identify novel pathway members or complex members, genes missed during the screening process, or genes that perform a specific function, such as protein kinases. STRING (https://string-db.org/) is a database of both experimentally verified and theoretically predicted interactions between proteins (Szklarczyk et al., 2021). These interactions occur through computational prediction, inter-organism information transmission, and aggregation of interactions from other (primary) databases, and they can be either direct (physical) or indirect (functional).
2.7 Effect of point mutation on the structural and functional integrity of the proteinThe formation of a protein complex is critical in controlling many biological activities. Therefore, different algorithms were employed to investigate the effect of Bcl-2G101V and Bcl-2F104L structural and functional properties. mCSM-PPI2 (http://biosig.unimelb.edu.au/mcsm_ppi2/) was used to predict the effects of missense mutations on protein-protein affinity (Rodrigues et al., 2019). mCSM-PPI2 uses graph-based structural signatures to model the effects of variations on the inter-residue interaction network, evolutionary information, complex network metrics, and energy terms to generate an optimised predictor. ConSurf (https://consurf.tau.ac.il/) is another tool we employed to estimate the evolutionary conservation of residue positions in a protein molecule based on the phylogenetic relationships between homologous sequences (Ashkenazy et al., 2016). The degree to which the residue position is evolutionarily conserved strongly depends on its structural and functional importance. The ConSurf value varied from 1 to 9, with one denoting residues with the least conservation and nine denoting residues with the most conservation. Other tools such as FTSite (https://ftsite.bu.edu/) (Ngan et al., 2012), HOPE (https://www3.cmbi.umcn.nl/hope/) and Stride (http://webclu.bio.wzw.tum.de/stride/) (Heinig and Frishman, 2004), were also used to provide deeper insight on the structural and functional integrity of the protein upon mutation.
2.8 Molecular dynamics (MD) simulations2.8.1 Systems preparationThe Protein Data Bank Repository (RCSB PDB) (https://www.rcsb.org/) provided a crystallized X-ray structure of the Bcl-2WT, Bcl-2G101V, and Bcl-2F104L with PDB entries of 6O0K, 6O0L, and 6O0M, respectively. The water molecules in the crystal structure were removed, and the missing hydrogen atoms were substituted for them, with the correct charges assigned at neutral pH. The Schrödinger suite’s Protein Preparation Wizard was employed for initial structure processing and energy minimization. To further reduce steric clashes between residues, we used the OPLS-2005 force field to minimize energy while setting the RMSD threshold to 0.30 for all structures (Shivakumar et al., 2012).
2.8.2 Molecular dynamics simulations and post-dynamic analysisMD simulations were carried out using AMBER18 software and its Particle Mesh Ewald Molecular Dynamics (PMEMD) module (Case et al., 2024; Darden et al., 1993). Protein systems were modelled, and atomic charges were assigned state using the standard Amber (FF14SB) force field within the Amber package. An in-house pdb4amber script was used to modify, rename, and protonate (histidine) Bcl-2 (Maier et al., 2015). The LEAP module was employed to generate Bcl-2 parameters and topology files. This was also used for system neutralization. Molecular minimisation was carried out using a constraint potential of 500 kcal/mol, with partial minimisation for 2,500 steps and full minimization taking 5,000 steps. Furthermore, a gradual heating from 0 to 310 K was implemented in the system. The unconstrained equilibration was performed for 5 ns while the atmospheric pressure was maintained at 1 bar with the help of a Berendsen barostat (Berendsen et al., 1984). Subsequently, production stages were conducted over 500 ns to understand the structural consequences of the mutations on Bcl-2.
The enzyme coordinates of Bcl-2WT, Bcl-2G101V, and Bcl-2F104L were saved every 1 ps, and their resultant trajectories were analysed using the AMBER18 integrated CPPTRAJ module (Roe and Cheatham, 2013). Post-MD analyses included root-mean-square deviation (RMSD), root-mean-square fluctuations (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), intramolecular hydrogen bonding, and dynamic cross-correlation matrix (DCCM). Furthermore, principal component analysis (PCA) was calculated to unravel the protein’s atomic displacement extent. The generated data and subsequent complexes were visualized using Microcal Origin analytical software (www.originlab.com), NMWiz implemented in Visual Molecular Dynamics (VMD) (https://www.ks.uiuc.edu/Research/vmd/) (Seifert, 2014; Humphrey et al., 1996).
3 ResultsThe Bcl-2 SNP dataset was obtained from the dbSNP and Ensembl databases. Approximately 52,619 variations in Bcl-2 have been identified, with 49,593 SNPs located in the intronic region, 163 SNPs classified as missense variants, 1,401 SNPs located in the 3′UTR area, 832 SNPs located in the 5′UTR region, and 115 synonymous variants, as reported by dbSNP and Ensembl. Missense mutations in the coding region were the current target of this study. As a result of further filtering to remove duplicate variations, 130 variants were selected for further investigation.
3.1 Sequence-based analysis of point mutationEight tools, namely, SIFT, PolyPhen2, CADD, REVEL, MetaLR, Mutation Assessor, FATHMM, and Predict-SNP were used to conduct sequence-based prediction and analyze the potential effects of Bcl-2 mutations. These eight tools separated deleterious mutations from tolerated ones (Supplementary Table S1). Out of 130 variants, SIFT and PolyPhen2 estimated 45 (∼35%) to be deleterious while CADD, REVEL, Mutation Assessor, FATHMM, and Predict-SNP predicted 19 (∼15%), 6 (∼5%), 30 (∼23%), 26 (∼20%), and 38 (∼29%) mutations as deleterious, respectively. However, the MetaLR algorithm predicted that all 130 (100%) variants were tolerated (Figure 2).
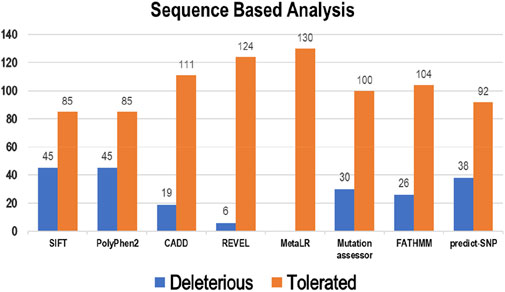
Figure 2. Deleterious and tolerated variations in Bcl-2 predicted through sequence-based algorithms.
3.2 Structure-based analysisMultiple computational algorithms, including mCSM, SDM2, DUET, PremPS, CUPSAT, ENCoM, MutPred-2, and DynaMut were used to provide structure-based predictions of the effect of mutations. These tools distinguished between destabilizing and stabilizing mutations (Supplementary Table S2). The analysis concluded that out of 130 mutations, mCSM: 120 (∼92%), SDM2: 85 (∼65%), DUET: 97 (∼75%), PremPS: 94 (∼72%), CUPSAT: 84 (∼65%), ENCoM: 60 (∼46%), MutPred: 2–27 (∼21%), and DynaMut: 61 (∼47%) mutations were estimated to be destabilizing the structure of the protein (Figure 3).
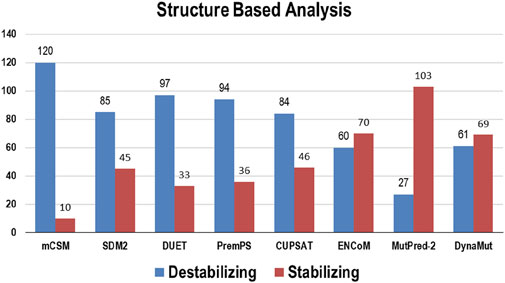
Figure 3. Destabilizing and stabilizing variations in Bcl-2 predicted through structure-based algorithms.
3.3 Disease phenotype analysisThe pathogenicity of the targeted mutations was assessed utilizing PhD-SNP, PANTHER, SNPs and GO, PMut, and Meta-SNP. These algorithms use their prediction values to determine whether a specific mutation is disease-causing or neutral. From the 130 mutations, PhD-SNP predicted 27 (∼21%) mutations to be pathogenic, while PANTHER, SNPs and GO, PMut, and Meta-SNP predicted 40 (∼31%), 20 (∼15%), 45 (∼35%), and 23 (∼18%) mutations associated with the disease, respectively (Figure 4). However, only 11 of these mutations were predicted to be disease-causing across all the prediction algorithms (R12G, V15L, H94P, L97P, R98L, R129P, G141E, V142G, N143S, M166T, and G193R) (Supplementary Table S3).
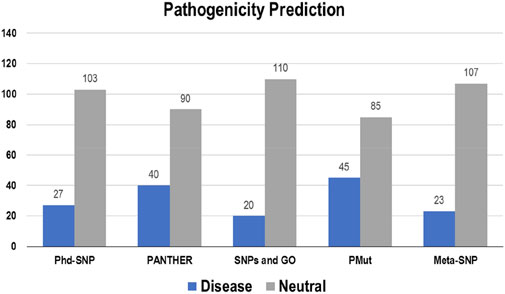
Figure 4. Disease and neutral variations in Bcl-2 predicted through disease phenotype prediction algorithms.
3.4 Post-transcriptional modification (PTM) sites predictionGPS-MSP 6.0 was used for methylation and determined the number of Bcl-2 sites that would be modified. However, GPS-MSP 6.0 predicted that phosphorylation would occur at 35 residues [Ser:15 (43%), Thr:12 (34%), and Tyr:8 (23%)]. In contrast, it was predicted by Netphose 3.1 those 20 different residues could be phosphorylated [Ser:11 (55%), Thr:7 (35%), and Tyr:2 (10%)].
Ubiquitination was predicted using BDMPUB and UbPred. BDMPUB anticipated that two lysine residues would be ubiquitinated, whereas UbPred projected those four lysine residues would be ubiquitinated.
3.5 Gene interaction networkThe interaction between Bcl-2 and other genes was evaluated using the GeneMANIA and STRING web servers. GeneMANIA analysis showed that Bcl-2 physically interacted with all ten genes and has no co-localization or genetic interaction with any other gene. However, Bcl-2 was co-expressed with BAX, BCL2L1, NLRP1, BBC3, and BID. Moreover, Bcl-2 shared protein domains with BCL2L1, BAX, BIK, and BID (Figure 5A).
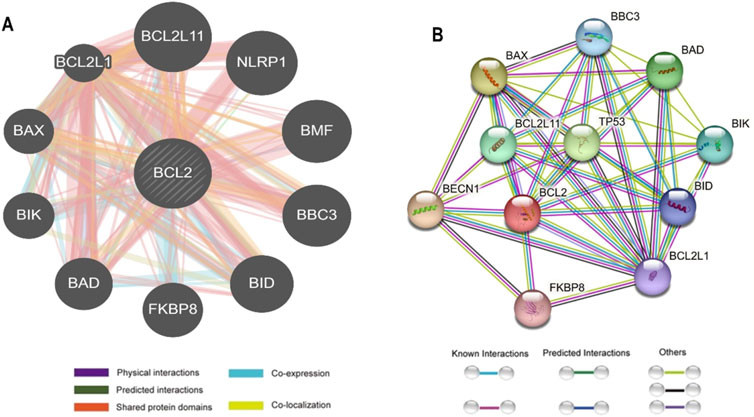
Figure 5. Bcl-2 Gene interactions with other genes predicted by (A) GeneMANIA and (B) STRING.
The STRING database offers an integrated and comprehensive evaluation of indirect (functional) and direct (physical) protein-protein interactions. The network analysis revealed that Bcl-2 interacted directly with BECN1, BAX, TP53, BAD, BCL2L11, BIK, BBC3, BID, BCL2L1, and FKBP8 (Figure 5B).
3.6 Effect of mutations on the structural and functional integrity of Bcl-23.6.1 Estimation of impact of mutation on protein-protein interactions (PPIs)The effect of mutations on the binding affinity of protein interactions was evaluated using mCSM-PPI2, which evaluates the effect of mutation by simulating the impact of variations on the network of non-covalent interactions between residues utilizing graph kernels, energetic terms, complex network metrics, and evolutionary data. The decreased binding affinity of protein-protein interaction was observed at the active site residues of the mCSM-PPI2-predicted Bcl-2 interaction, with a change in affinity (ΔΔGaffinity) of −0.559 kcal/mol for the G101V variant and −1.053 kcal/mol for the F104L variant. The interaction network revealed that the wild-type protein residue Gly101 established hydrogen bonds with Tyr18, Leu97, Arg98, Phe104, and Ser105, as well as van der Waals interactions with Gln99 and Glu152; however, in the mutant, Val101 established a hydrogen bond with Leu97, Arg98, Phe104, Ser105, and Glu152 (Figure 6). Likewise, the Phe104 in the wild-type generated hydrogen bonds with Ala100, Gly101, and Tyr108, and van der Waals interactions with Ala100, Asp102, Arg106, Tyr108, and Phe123, while in the mutant, leucine formed hydrogen bonds with the same residues in the wild-type (Figure 6).
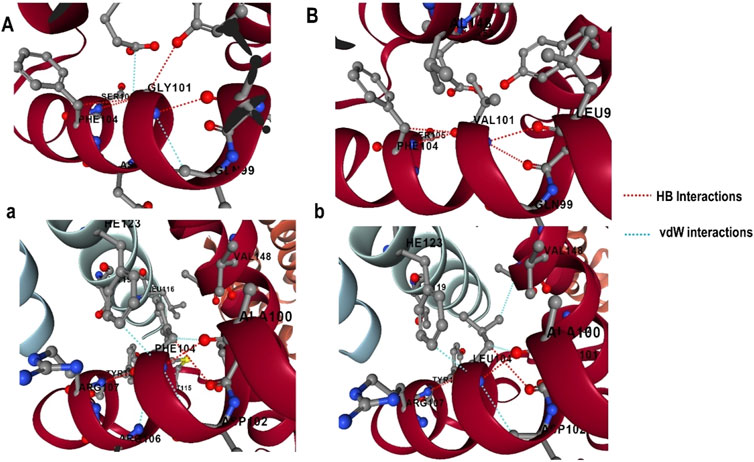
Figure 6. G101 and F104 residue interactions network of Bcl-2; (A) wild G101, (A) G101V variant, (B) wild F104, and (B) F104L variant as predicted by mCSM-PPI2.
3.6.2 Conservation analysis of Bcl-2The conservation of residues is the primary factor that ensures the structural integrity of proteins. The Bcl-2 structure’s conservation of residues was investigated using the ConSurf web server to comprehend its significance and localized evolution. The arrangement of residues and their degree of conservation was uncovered utilizing the ConSurf analysis. Several residues in Bcl-2 were shown to be relatively conserved using ConSurf, with particular emphasis on G101 and F104, suggesting that genetic variations at these positions might substantially impact Bcl-2 (Figure 7).
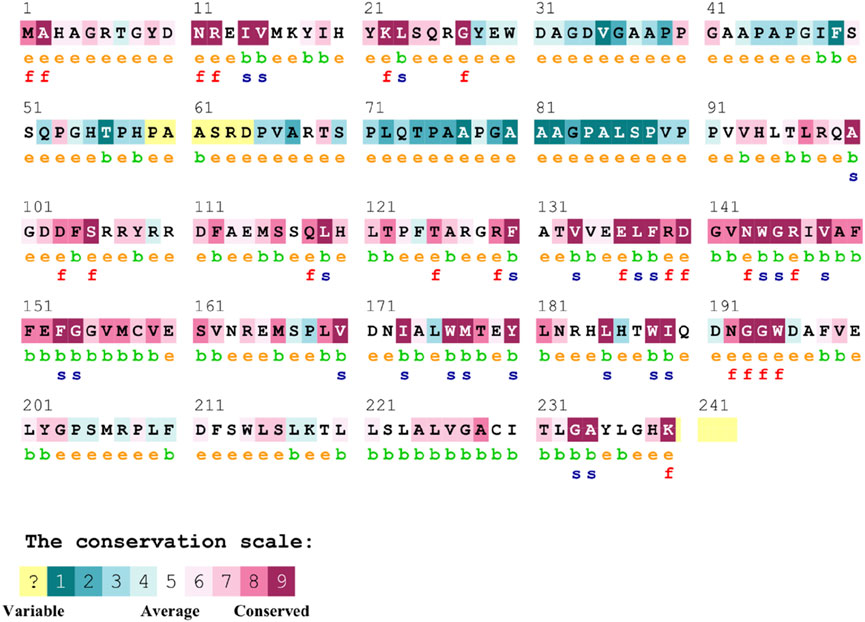
Figure 7. Sequence conservation plot of Bcl-2 protein generated using ConSurf web server.
3.6.3 Mapping ligand binding sites of Bcl-2The FT-site web server was used to identify Bcl-2 binding sites based on experimental evidence. The FT-site server depicted three ligand sites in Bcl-2. The ligand sites in Bcl-2 were represented by three different mesh-like structures on the FT-site server (pink, green, and purple), with corresponding residues that are within 5.0 Å of the binding site represented by ball and stick in these sites (Figure 8). The position of the F104 residue is detected in the first and second ligand-binding sites, while G101 is detected in the second ligand-binding site (Table 1). Consequently, mutations G101V and F104L may be more deleterious, as they potentially impact the Bcl-2 ligand-binding affinity.
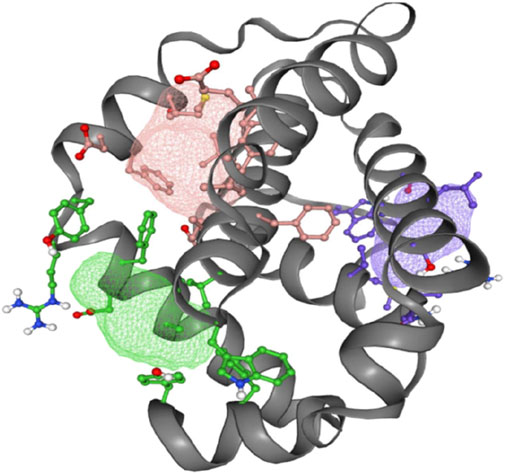
Figure 8. FT-site server prediction of the Bcl-2 protein ligand binding sites represented in mesh-like structure: pink (binding site 1), green (binding site 2), and purple (binding site 3).

Table 1. Bcl-2 protein ligand-binding sites and their respective residues.
The HOPE project PDB viewer was used to visualize the structural features of the Bcl-2WT, Bcl-2G101V, and Bcl-2F104L (Figure 9). Each residue demonstrated a unique size, charge, and hydrophobicity. These values frequently varied between the original wild-type and the newly introduced mutant residues. For the Bcl-2G101V, the mutant residue was bigger and more hydrophobic than the Bcl-2WT residue. Although the mutated residue is not directly involved in ligand binding, it may indirectly affect ligand interactions made by other residues due to changes in local stability. The mutated residue is located within a special BH3 motif. Therefore, the different properties of residues caused the motif to become disrupted and consequently impair its function. Glycine had the highest degree of flexibility compared to other residues, which may be necessary for protein function. This function can be abolished by mutating this glycine. For Bcl-2F104L, the mutant residue was smaller than the Bcl-2WT residue. The Bcl-2WT residue interacted with Venetoclax, and the difference in properties between the Bcl-2WT and mutant can easily cause a loss of interactions with the ligand. Protein function was frequently dependent on ligand binding, and this mutation may impair this function. The mutated residue was located within a special BH3 motif near a highly conserved position. Consequently, the motif was disturbed owing to the different properties of the residues, which would impede its function.
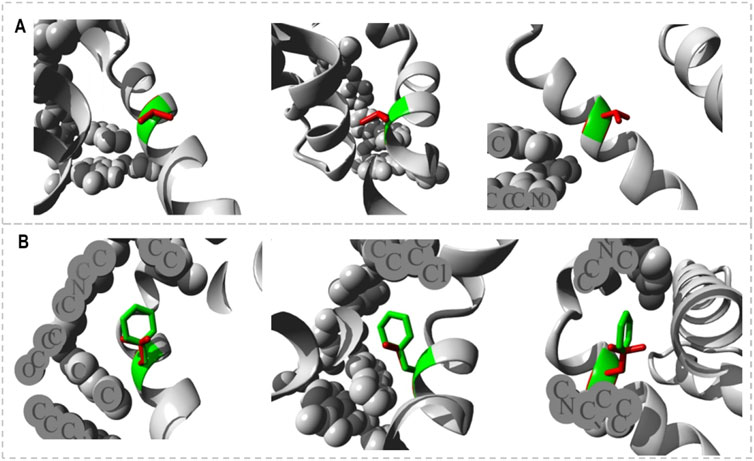
Figure 9. Close-ups (different angles) of the mutant and wild system; (A) Bcl-2G101V and (B) Bcl-2F104L.
3.6.4 Investigating the effect of the mutations on the protein secondary structureMD trajectories of 500 ns were used to investigate the dynamics of secondary structural elements in Bcl-2WT, Bcl-2G101V, and Bcl-2F104L. This study contributed to a better understanding of the effects of genetic variations on the Bcl-2’s secondary structure through simulations. The STRIDE web server was used to detect the change in secondary structure at 10, 100, 200, 300, 400, and 500 ns (Figure 10). The secondary structural components in Bcl-2, such as α-helix, 3–10 helix, and turns, were divided into specific residues at each time interval. The Bcl-2G101V and Bcl-2F104L were observed to switch from a helix to a turn configuration at these residues.

Figure 10. The secondary structural analysis of the Bcl-2WT, Bcl-2G101V, and Bcl-2F104L at 10, 100, 200, 300, 400, and 500 ns using the STRIDE web server.
3.7 Dynamic and conformational stability and fluctuationsThe inherent behavior of a protein is associated with conformational changes and structural aberrations. Modifying a protein’s structure can significantly affect its function]. Therefore, understanding mutation-induced structural changes requires a more in-depth investigation of the conformational dynamics of proteins. For this reason, the effects of Bcl-2 mutations (G101V and F104L) were investigated over 500 ns MD simulations. The dynamics and stability of Bcl-2WT, Bcl-2G101V, and Bcl-2F104L were determined by evaluating the time variable considering the RMSD of Cα atoms from computed trajectories. All systems reached convergence after 100 ns of the simulation period (Figure 11A). The Bcl-2WT exhibited the lowest deviated RMSD value, 1.14 Å, while the Bcl-2G101V and Bcl-2F104L revealed higher RMSD values, 1.43 and 1.62 Å, respectively. The Bcl-2G101V disrupted the RMSD pattern of Bcl-2WT and caused it to fluctuate more than the Bcl-2WT and Bcl-2F104L during the simulation. The findings showed that Bcl-2WT and Bcl-2F104L displayed the least deviation of Cα atoms compared to Bcl-2G101V, indicating that the mutation of Gly to Val reduced the structural stability of Bcl-2. Furthermore, no significant variations in structural snaps were noticed, excluding the α3-α4 helices (hydrophobic groove) of superimposed Bcl-2WT, Bcl-2G101V, and Bcl-2F104L every 100 ns during the simulation (Supplementary Figure S1). Here, α3-α4 helices become more dynamic and flexible as the simulation progresses, thus inducing expansion or shrinking in the hydrophobic groove, which appears most effectively in the Bcl-2G101V.
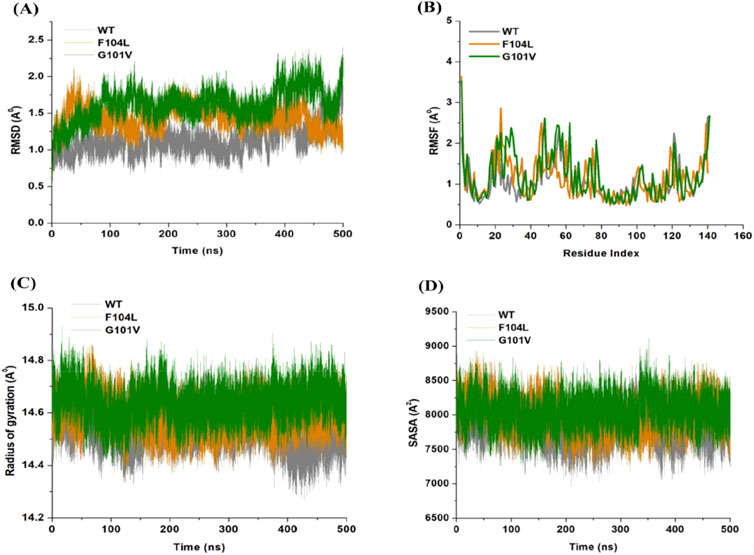
Figure 11. (A) RMSD, (B) RMSF, (C) Rg, and (D) SASA values across Cα of Bcl-2WT (gray), Bcl-2F104L (orange), and Bcl-2G101V (green) over 500 ns MD simulations.
The relative rigidity and flexibility of residues determined protein conformational changes and their associated functions. Consequently, the RMSF values of Bcl-2WT, Bcl-2G101V, and Bcl-2F104L can be computed and analyzed to see how Bcl-2’s residual fluctuations change due to mutations (Figure 11B). Bcl-2WT demonstrated the least fluctuations of the residues with an average RMSF value of 1.10 Å when compared to 1.13 and 1.20 Å for the Bcl-2F104L and Bcl-2G101V, respectively. The calculated trajectory showed a slightly higher pattern of fluctuations, especially for the Bcl-2G101V variant. As a result of these mutations, the regions surrounding the various sites become more dynamic and internally disturbed, reflecting higher fluctuations in Bcl-2. The RMSF distribution correlated with the RMSD pattern, with mutated systems exhibiting more significant fluctuations. The substantial variations in the mutants’ residual fluctuations could be attributed to Bcl-2 structural inactivation.
Furthermore, the Rg values of all three systems were analyzed to determine the folding behavior and overall conformational changes in the Bcl-2 structure before and after mutation induction. The compactness, stability, and folding of a protein can be determined from the change in Rg values over time. The Rg values of the Bcl-2WT, Bcl-2G101V, and Bcl-2F104L were estimated from the MD trajectories and plotted (Figure 11C). Bcl-2WT had the lowest Rg value (14.54 Å, while the Bcl-2F104L and Bcl-2G101V showed slight increases at 14.59 and 14.63 Å, respectively. Altogether, Rg analysis of Bcl-2 revealed that the mutants were less stable, more flexible, and less compact than the native protein.
Moreover, the Bcl-2 structure’s hydrophilic and hydrophobic residues were analyzed using SASA. The SASA values for the Bcl-2WT, Bcl-2G101V, and Bcl-2F104L were obtained and plotted throughout the 500 ns of MD simulation (Figure 11D). Following exposing the system to the solvent, Bcl-2WT had a median SASA value of 7,824 Å2. The Bcl-2G101V exhibited a higher SASA value of 8,049 Å2 than that of the Bcl-2F104L, which displayed a value of 7,985 Å2. The SASA values of all three systems agreed with the Rg results. The differences in the SASA values for the three systems throughout the simulation reflect Bcl-2 unfolding and folding. The overall SASA values for Bcl-2WT and Bcl-2F104L were slightly different, suggesting that the structural mutation from Phenylalanine to Leucine at position 104 in Bcl-2 provides better exposure to solvent compared with Bcl-2G101V and, thus, favors the enhanced activity of the Bcl-2F104L over that of the Bcl-2G101V.
3.7.1 Hydrogen bonding analysisAnalysis of intramolecular hydrogen bonds primarily assists in evaluating the overall conformation and stability of the protein structure through MD simulations. Time-dependent intramolecular hydrogen bond analysis was performed and plotted to evaluate the effect of mutations on the structure of Bcl-2 (Figure 12). The average values of intramolecular hydrogen bonds in Bcl-2WT, Bcl-2G101V, and Bcl-2F104L ranged from about (43–100), (41–98), and (40–96), respectively, indicating a slight change before and after mutation formation. The Bcl-2F104L and Bcl-2WT models were more compact and stable than the Bcl-2G101V model, and the results maintained a roughly similar trajectory pattern.
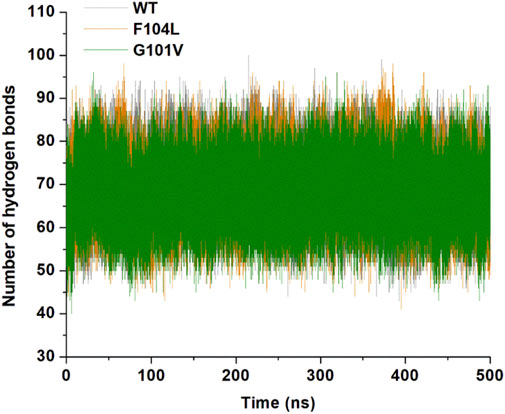
Figure 12. Intramolecular hydrogen bonding in Bcl-2WT (gray), Bcl-2F104L (orange), and Bcl-2G101V (green) over 500 ns MD simulations.
3.7.2 Dynamic cross-correlation matrix (DCCM)To examine the differences in the dynamics of Bcl-2WT, Bcl-2G101V, and Bcl-2F104L, DCCM plots were generated for anti-correlated and correlated protein structural motions. The residues’ motion values range from −1 to +1. Positive values indicate positively correlated motions (brown colour), whereas negative values indicate anticorrelated motions (black colour) between residues (Figure 13). The scatter plots revealed that motion modes between residues of Bcl-2F104L are similar to those of Bcl-2WT, whereas the Bcl-2G101V showed a slightly different pattern, mutation obviously enhances the positively correlated motions occurring in the Bcl-2.
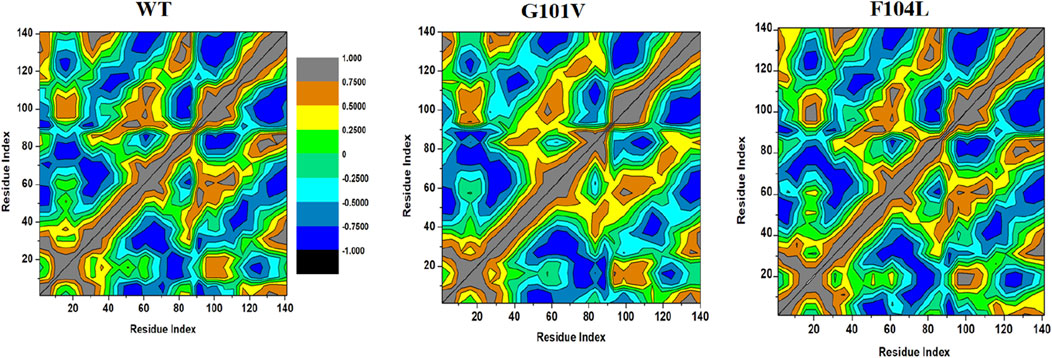
Figure 13. DCCM analyses for Bcl-2WT, Bcl-2G101V, and Bcl-2F104L over 500 ns MD simulations.
3.7.3 Principal component analysis (PCA)Intensive movements in Bcl-2WT, Bcl-2G101V, and Bcl-2F104L were evaluated using PC analysis with the first two eigenvectors (EVs) to qualitatively examine the influence of induced mutations on the major conformational movements of each residue (Kumalo et al., 2016). The eigenvectors illustrate the directions of Bcl-2 motion, and the eigenvalues represent the overall motion strength; these are obtained by diagonalizing the covariance matrix (Chen et al., 2021; Chen et al., 2022). The conformational changes of Bcl-2 and its variants were shown in a 2D scatter plot (Figure 14), indicating a significant change in Bcl-2 overall movements after acquiring the mutations, especially Bcl-2G101V. Moreover, Figure 14 shows that the Bcl-2G101V and Bcl-2F104L with the trace covariance matric of 12.46 and 22.46 Å2, respectively, imposed highly fluctuated anti-correlated effects as the negative values of 2D scatter point into the protein. In the case of Bcl-2WT, the trace covariance matrices were 24.09 Å2, indicating the presence of prominent correlated motions with minimal system fluctuations. Consequently, the findings demonstrated that the Bcl-2G101V caused substantial fluctuations in the simulated Bcl-2 dynamics.
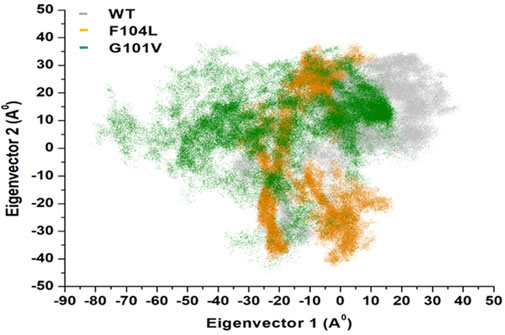
Figure 14. PCA for Bcl-2WT (gray), Bcl-2F104L (orange), and Bcl-2G101V (green) over the 500ns MD simulations.
4 Discussions4.1 Sequence, structure, phenotype-mutational analysis and gene interactionsTo ascertain the deleterious effect of residue mutation on the protein, we employed various sequence-based point mutation algorithms. Out of 130 mutations, SIFT and PolyPhen2 algorithms displayed the highest estimation, deeming 45 mutations (∼35%) deleterious. With the exception of the MetaLR algorithm, which predicted that all 130 (100%) variants were tolerated, other algorithms displayed results ranging from around 5 to 23 percent (Figure 2). We hypothesize that the inclusion of machine learning and high-throughput Next-Generation Sequencing (NGS) data in the PolyPhen2 method broadened the search field, contributing to the high prediction rate. Similarly, various algorithms were adopted to predict the effect of missense mutations on the protein stability, and to distinguish between destabilizing and stabilizing mutations. Out of 130 mutations, 3 algorithms (ENCoM, MutPred, and DynaMut) assessed b
留言 (0)