
記住我
Globally, colorectal cancer (CRC) is the fourth most common cancer to be diagnosed and the third most common cause of death from cancer (1, 2). Each year, around 250,000 cases of CRC are identified in Europe, making up 9% of all malignancies. The incidence is slightly greater in Western and Northern Europe compared to Southern and Eastern Europe (3). Its causes are complex and multifaceted, inherited in only 5% of cases (4). Non-dietary risk factors for colorectal cancer include smoking and prolonged use of drugs like non-steroidal anti-inflammatory drugs (NSAIDs) and aspirin (5). Two screening methods are available, fecal occult blood test (FOBT) and endoscopy. Randomized studies have shown that FOBT can reduce the death rate of colorectal cancer by up to 25% in those who attend at least one screening session (6). Surgery is the primary therapy option for people with likely curable colorectal cancer. This is usually accompanied by adjuvant therapy, a systemic treatment that lowers the risk of recurrence and mortality. Additionally, pathological staging can be used to predict the recurrence rate (3). Activated oncogenes and inactivated tumor suppressor genes play crucial roles in various phases of colorectal cancer development (7). However, single-gene regulation systems cannot account for all characteristics of malignant behavior or be responsible for every cancer marker (8).
The discovery of altered proteins or metabolites during the progression of colorectal cancer is crucial in identifying novel potential biomarkers for early detection (9). Proteomics is a comprehensive and advanced methodology characterized by its ability to detect thousands of proteins simultaneously in various sample types, including cells, tissues, and bodily fluids. Research into specific biomarkers (10) and therapy pathways can greatly improve patient outcomes (11). Deciphering the underlying molecular processes and regulatory networks (12) of identified biomarkers in colorectal cancer remains an important challenge (13). For example, 5-aminovalerate interacts with the bacterial species Adlercreutzia, as does cholesteryl ester with Blautia, Roseburia, and Staphylococcus. Numerous genes have also been implicated in processes specific to epithelial cells, notably with the oxidative phosphorylation pathway and associated genes, which indirectly control cholesterol esterification in colorectal cancer (14).
Understanding the biology of cancer requires a thorough understanding of protein expression changes and interactions, which proteomics (15) provides. It has been established that proteins play a significant role in the development of biomarkers and pharmaceutical targets (16) and may be used as a window into human health (17). A comprehensive study identified 7526 proteins by label-free quantitative proteomics of 64 colon cancer tissues and 31 rectum cancer tissues are among the most comprehensive to date (18). Proteomic studies have made it easier to identify protein targets and signaling pathways involved in the development of cancer. For example, some of the proteins identified were involved in IL-17 signaling pathways in colorectal cancer progression (19). Comprehensive combined proteomic and genomic studies of CRC have been completed, leading to the discovery of treatment targets, cancer antigens, CRC subtypes, and critical signaling pathways associated with the progression of CRC (20). Even though there has been a substantial amount of research conducted on biomarkers for primary colorectal cancer, clinical guidelines in both the USA and Europe, such as those provided by the National Comprehensive Cancer Network and European Society for Medical Oncology, currently prioritize tumor-node-metastasis staging and the identification of DNA mismatch repair deficiency or microsatellite instability (21). These guidelines also consider traditional clinicopathological factors when making a diagnosis (22).
Breakthroughs in omics technologies, such as RNA sequencing (23) for transcriptome gene expression profiling, next-generation sequencing (NGS) (24), and mass spectrometry (25) have facilitated the use of molecular markers in diagnosing colorectal cancer (26). The integration of healthcare characteristics like clinical data, gene expression, and miRNA expression with machine learning and AI (27) based methods can maximize the utilization of omics data (28). Molecular interactions and biomarkers were discovered through hierarchical clustering, protein-protein networks, and correlation analyses (29). Proteomic signatures, which offer a tool for forecasting CRC stages and identifying biomarkers, were constructed by selecting differentially expressed proteins (DEPs) using Least Absolute Shrinkage and Selection Operator (LASSO) and Support vector machine (SVM) (30). Sensitive diagnostic tools to detect cancer were found using machine learning algorithms like SVM, random forest, and decision trees (31). Other non-omics methods have been used such as nanotechnology (32) and imaging techniques to better understand the disease (33). Despite the advancements made in understanding the molecular characteristics, biological markers, and therapeutic targets of colorectal cancer, the complexity of its biology, severe outcomes, and extensive metastasis highlights the need for additional research in identifying predictive and prognostic biomarkers (34).
Here, we present the comprehensive analysis of the Olink-based quantitative proteomics in UK Biobank data. With the application of Explainable artificial intelligence (XAI) models and validation of models with the Bosch et al. dataset, our work aims to identify protein biomarkers and their mechanisms that may be employed as diagnostic markers across a range of proteomic datasets. Ultimately, these discoveries might lead to improved cancer detection and the emergence of greater accuracy models.
2 Materials and methods2.1 Study design and participantsThe study was carried out in two phases, initially using proteomics data sourced from UK Biobank, with the cohort in Bosch et al. as a validation dataset. The UK Biobank is a large-scale cohort research project, consisting of 500,000 individuals recruited between 2006 and 2010 from various sites around the UK, aged 40 to 69.
Colorectal cancer patients were first identified using diagnosis information from various sources of UK Biobank, with all details and diagnosis codes detailed in Supplementary Table S1. Then participants with age and sex were matched to participants with no ICD-10 diagnosis in their inpatient data using the MatchIt package. Participants with metabolite information (as of July 2021) and proteomic data (as of July 2023) were followed up for analysis.
We conducted our comprehensive proteomics data analysis using two datasets which include one from the UK Biobank and another from Bosch et al. UK Biobank is used as training and Bosch et al. used as a validation dataset. The details of the datasets used in this study are summarized in Table 1. In the first phase,159 significant proteins of UK Biobank were used to validate with Bosch et al. data, and in the second phase we used 98 proteins that were common among both datasets to validate (Figure 1). For UK Biobank, proteomics data was generated using the antibody-based Olink Explore 3072 PEA (35) and for Bosch et al. (36) proteins were generated using liquid chromatography-mass spectrometry (LC-MS).
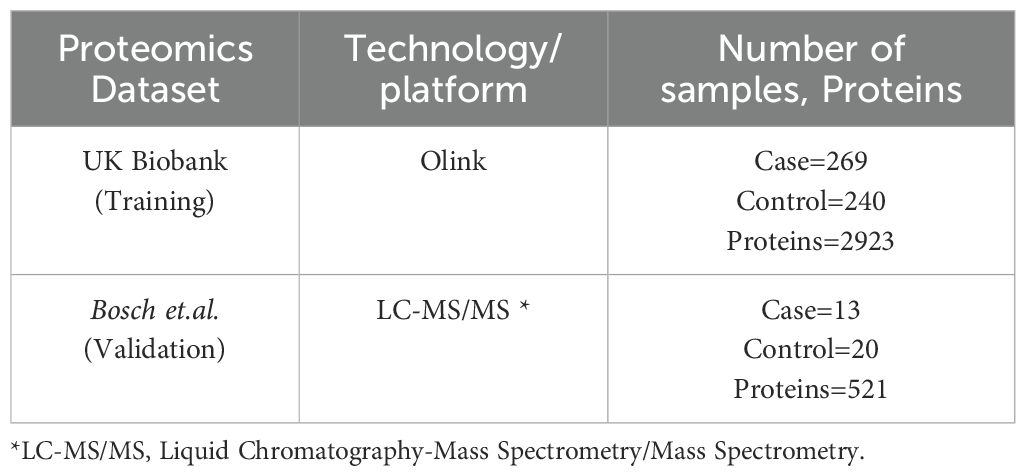
Table 1. Datasets used in the study.
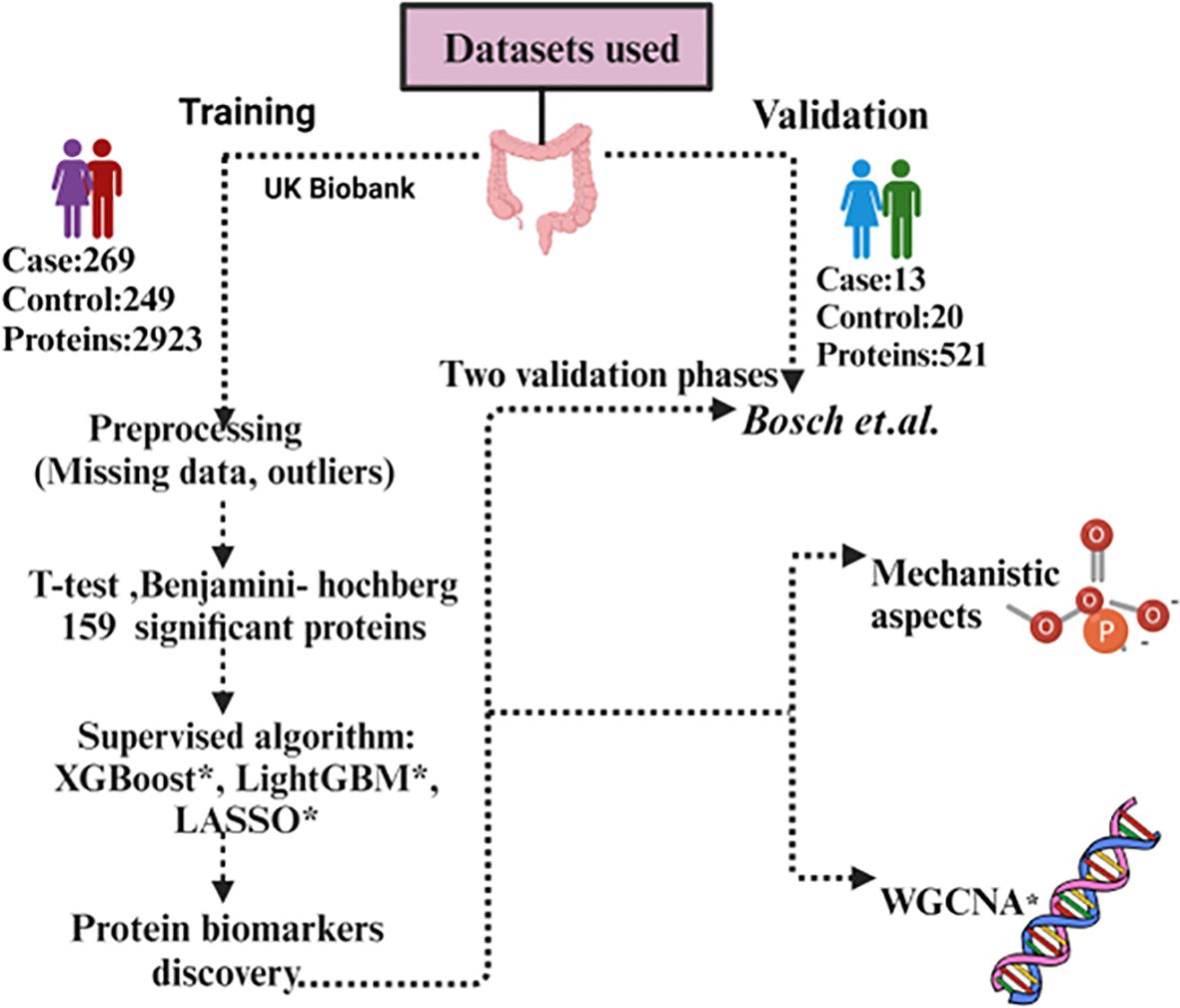
Figure 1. Workflow of the data analysis using UK Biobank and Bosch et al. dataset. *XGBoost, eXtreme Gradient Boosting; *LASSO, Least Absolute Shrinkage and Selection Operator; *LightGBM, Light Gradient-Boosting; *WGCNA, Weighted gene co-expression network analysis.
2.2 UK Biobank data processingAll pre-processing steps are performed using R (v4.2.0). Imputation of 20% missing value protein which has 1460 proteins (37) was done.
We used several methods mean, median, Classification and regression trees (CART), and K-Nearest Neighbors (KNN) for imputation to compare the distribution before and after imputation (Supplementary Figures S1A–E). The mean imputed dataset had a close relation to the original dataset distribution. Kolmogorov Smirnov Test (K-S test) is also performed to find the statistical difference among different imputation methods. Mahalanobis distance (38) has been used for outlier detection based on the 99th percentile threshold using the mean and covariance matrix of our data. It was determined that the dataset’s threshold was 22.71. Six samples were found to be outliers (Supplementary Figure S2) and discarded from the study. For further statistical analysis and machine learning tasks, the pre-processed dataset (1460 proteins and 512 samples) was used.
2.3 Machine learning modelIn this study, we used three machine learning models, eXtreme Gradient Boosting (XGBoost) (39). Light Gradient-Boosting Machine (LightGBM) (40), and Least Absolute Shrinkage and Selection Operator (LASSO). XGBoost and LightGBM make use of an ensemble of classification trees and combine their prediction from multiple individual decision trees to make more accurate and robust predictions, LASSO on the other hand, it is a regularization algorithm that helps reduce the feature space, highlighting key association. All machine learning analysis were carried out in python (v3.9.13) “scikit learn” module (v1.0.2).
For this study, the selection of LASSO, XGBoost, and LightGBM is based on several aspects. With strong feature selection and regularization capabilities, LASSO regression is especially well-suited for high-dimensional datasets, which are frequently employed in biomedical research. By implementing a penalty, LASSO reduces overfitting and efficiently selects the most pertinent predictors by shrinking less significant coefficients to zero (41). Research has shown that LASSO is useful for finding important risk variables and biomarkers in cancer studies. The interpretability and accuracy of predictive models were enhanced when Ma et al. (42) effectively used LASSO to choose prognostic characteristics in colorectal cancer. XGBoost is well-known for its high Area Under the Curve (AUC) scores in classification tasks, its ability to handle missing data and non-linear interactions (39), and its successful application in predicting the survival of colorectal cancer (43). LightGBM is perfect for large-scale datasets because it is scalable and fast, manages high-dimensional, sparse data effectively, and provides superior AUC (40). Recent research, including Zhang et al. (44) and Onwuka et al. (27), supports the application of these models by showing that they may accurately and clinically meaningfully identify the relationships between variables in colorectal cancer. The study utilized SHapley Additive exPlanations (SHAP) to enhance the interpretability of the machine learning models, addressing a key limitation of complex algorithms like XGBoost and LightGBM—their “black-box” nature. Even though these models are quite effective and precise, it is important to comprehend their predictions in a clinically useful way, particularly in colorectal cancer research where it is critical to identify important risk variables and how they affect the result. By quantifying each feature’s contribution to the model’s output, SHAP offers a cohesive framework for explaining individual predictions (45). By using SHAP, the study makes sure that the high AUC scores achieved by the machine learning models are accompanied by interpretability and transparency, which makes the results useful for clinical decision-making. The study’s objective of identifying important risk variables and their impact on the outcomes of colorectal cancer is also in line with SHAP’s ability to identify feature relevance at both the global (model-wide) and local (individual prediction) levels. Each of the methods has complementary properties. LASSO is a linear method and able to perform sparse feature selection, by shrinking the coefficients of less important features to exactly zero and hence captures the most comfortable linear relationships. XGBoost and LightGBM, however, are more versatile, and capable of capturing complex, non-linear relationships in the data. LightGBM is able to capture model building for categorical features. Together with SHAP for interpretability, these three models—LASSO, XGBoost, and LightGBM—complement one another by fusing their specific strengths to provide a robust and clear analysis. LASSO helps by regularizing the model, decreasing overfitting, and enhancing interpretability by efficiently managing high-dimensional data. This aids in the model’s simplification, making it easier to handle and understand. In contrast, XGBoost (39) and LightGBM) (40), are strong boosting algorithms that offer high predictive accuracy, both of which are important for producing accurate predictions. SHAP depicts how each feature affects the model’s predictions to give an understanding of feature relevance.
2.4 Hyperparameter optimizationThe proteomic dataset underwent an initial 80%-20% train and test split (27). We employed the GridSearchCV (46) method to determine the most effective values for each model’s hyperparameters. GridSearchCV is a utility within the Scikit-learn library designed for iterating through predetermined hyperparameters and training the model on the provided dataset. Stratified k-fold cross-validation with 5-fold was employed within grid search ensuring robust model assessment and hyper-parameter tuning by assessing all possible combinations of specified parameter values. The accuracy metrics collected for each hyperparameter combination were used to decide on top-performing models.
2.5 Machine learning model evaluationThe study utilized the UK Biobank dataset for training and testing and the data from Bosch et al. for validation of the performance of three distinct machine learning models (LASSO, LightGBM, and XGBoost). Each model underwent evaluation using various metrics including precision, recall (47), specificity (48), F1 score, and AUC-ROC (49). We incorporated a validation strategy to assess the quality of our findings and test the robustness of the proteins found in our model. Our experimental methodology utilized the k-fold cross-validation technique (k=5), a widely accepted approach (50). The model’s effectiveness was assessed by calculating the average performance metrics across 50 iterations (Supplementary Figure S3).
2.6 Feature selectionWe used SHapley Additive exPlanations (SHAP) for feature selection. SHAP quantifies the significance of each characteristic by utilizing ideas from cooperative game theory to explain how each protein contributes to the model’s predictions. This method improves the model’s interpretive skills while also offering insights into the decision-making process, which raises the output of the model’s transparency (51). This was visualized using a SHAP global importance plot to identify the proteins that had the greatest influence. Additionally, plots were used to show the relative contributions of each top protein to the sample. Finally, the common proteins of both datasets were visualized using a heatmap.
2.7 Statistical analysesThe present study used R (v4.3.0) for all pre-processing analyses of the UK Biobank, while Python (v3.9.13) was used for machine learning and interpretable XAI-based modeling. The Benjamini-Hochberg principle (52) was used to adjust p-values for multiple testing corrections, with values that fell below the critical false discovery rate (FDR) of 0.05 deemed significant. When dividing the datasets between sets for testing, training, and cross-fold validation for the machine learning tasks, they were stratified according to the target class. The mean AUC score across the folds together with the 95% confidence interval in Python provided a representation of the models.
2.8 Knowledge integration using STRING databaseThe protein–protein association network is one of the most efficient, wide-ranging forms of networks, it includes every gene that codes for a protein in a particular genome and depicts the functional interactions between those genes (53). Protein-protein interactions that are known or predicted are documented in the STRING (54) database. We compiled the list of proteins from both phases and uploaded it to STRING for analysis with a confidence interval of 0.4 classified as ‘medium confidence’ for predicted interactions and obtained a comprehensive view of potential interactions.
2.9 Human protein atlasTo understand the dynamic expression of protein-coding genes and to generate a map of the human proteome, the Human Protein Atlas (HPA) project was initiated as a part of the Human Proteome Project (HPP) focusing on antibody-based proteomics and integrated omics (55). The objective of HPA is to identify the expression and spatial distribution of each human protein in various human organs, cancer types, and cell lines. We have employed HPA to get more knowledge of the significant role that proteins play in the many types of cancer cells.
2.10 Weighted gene co-expression network analysisWe performed a Weighted Gene Co-expression Network Analysis (WGCNA) (56) on both of our datasets to identify modules of co-expressed genes and their association with clinical traits, specifically focusing on cancer and non-cancer control samples. The analysis began with the selection of an optimal soft-thresholding power (β) to ensure the network’s scale-free topology.
Next, a module detection test was carried out via the hierarchical clustering method to identify distinct modules with co-expressed genes. After identifying distinct modules, we performed a correlation matrix with cancerous and non-cancerous samples to indicate genes that are upregulated or downregulated in diseased cases. The list of upregulated or downregulated genes was assessed for functional annotations from the Kyoto Encyclopedia of Genes and Genomes (KEGG) via pathway enrichment analysis to identify biological pathways that can serve as potential therapeutic targets.
2.11 Protein quantitative trait loci analysisProteomic analysis was carried out for seven proteins (AHCY, CEACAM5, LCN2, RETN, SELE, TFF1, and TFF3) in the UK Biobank proteomics data against imputed SNP array data (57). The cohort was split into controls without CRC, those who had CRC before the blood sample was taken, and those who received a diagnosis afterward (numbers depend on missingness of proteomics data but a maximum of 51002, 332, and 884 respectively). Analysis was carried out using regenie (58) v3.4.1, with covariates including sex, age, age2, age x sex, age2 x sex, genetic principal components 1-10, and genetic array used. Sex was coded as 1 for females and 2 for males. Gene annotations were applied using biomaRt (59) v2.56.1, with the dataset hsapiens_gene_ensembl for GRCh37.
2.12 Batch-effect correctionThe common proteins between the UK Biobank and Bosch et al. datasets were combined for ComBat analysis (60). Batch effects (61) are expected to have an impact on the integrated dataset due to the variations in cohort characteristics and data-collecting techniques. Using the R package “sva,” (62) we implemented the ComBat batch-effect correction method to resolve these disparities. This method adjusted for systematic differences while maintaining significant biological diversity, harmonizing the protein expression data across datasets. This stage made sure that the source of the dataset wouldn’t affect subsequent analysis. After batch-effect correction, batch corrected dataset was split for testing (20%) and training (80%). We used a 5-fold cross-validation technique in the training phase to ensure a reliable and unbiased assessment of predicted performance. Three models for prediction were used LASSO regression, LightGBM, and XGBoost. To optimize performance, hyperparameter optimization was done using grid search. Each model’s performance metrics, such as accuracy, ROC-AUC, precision, recall, and F1-score, were computed in order to thoroughly assess how well it predicted the variable being studied. Further we analyzed the potential biomarkers that were found to be particularly important to the desired outcome, in addition to assessing the models on the entire dataset, and the same performance measures were used to evaluate each model’s predictive value. This provided us with information about the relative significance and predictive ability of these particular proteins by comparing the performance of the entire dataset with those on individual biomarkers. To evaluate feature importance and determine which feature had the most influence on model predictions, potential proteins were then put through SHAP analyses. A better comprehension of the variables influencing the models’ outputs and their conformity to biological relevance was made possible by the results of this study.
3 Results3.1 Dataset cohortAfter extracting participants in UK Biobank with a colorectal diagnosis (as defined in Supplementary Table S1), 9,890 participants were identified, with 6372 having a colorectal diagnosis coming from more than 1 source and 3518 coming from just a unique source. From those unique sources, death registries identified 74 participants, cancer registries 325, hospital inpatient data 2737, and self-reported information 382. These 9,890 participants were then age and sex-matched to controls (as defined by having no ICD-10 diagnosis recorded in hospital inpatient data) generating a dataset of 19784 participants. After filtering for those with metabolite information (as of July 2021) and protein data (as of July 2023), 509 participants had all information remaining, corresponding to 269 cancer patients and 240 controls. 5 participants had extra protein follow-up information, yielding a total of 518 samples with protein information used. Basic demographic information can be found in Table 2.
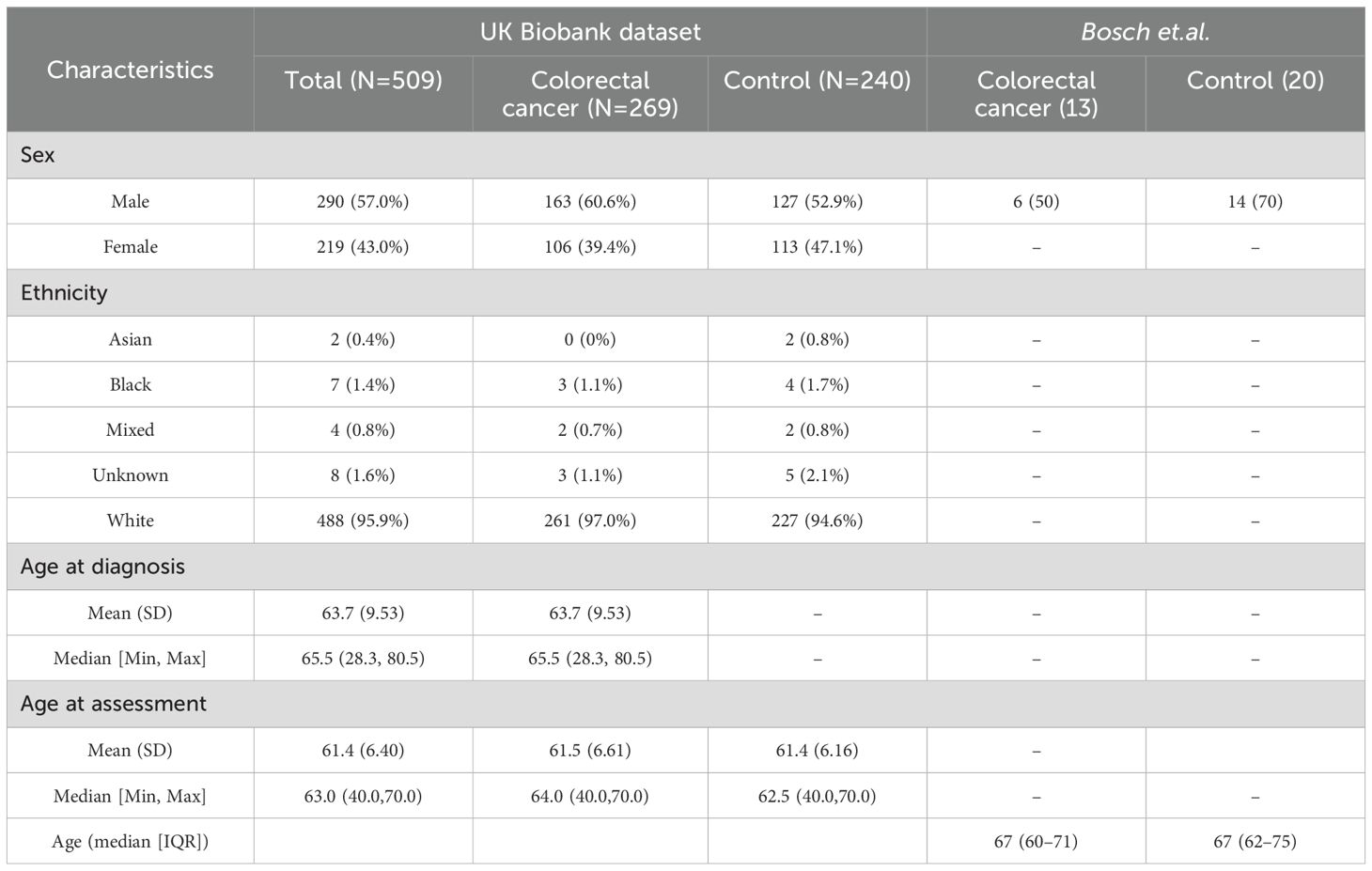
Table 2. Baseline demographic characteristics.
Regarding our validation dataset, Bosch et al. had 33 participants (13 cancer patients and 20 controls) with a median age of 67 among colorectal cancer participants which included 6 male participants (Table 2). For the downstream analysis, 518 samples and 2923 proteins were assessed for UK Biobank, and 33 samples with 521 proteins made up the validation dataset Bosch et.al.
3.2 Univariate analysisUnivariate comparisons between cases and controls found 159 significant proteins (FDR<0.05) in UK Biobank and 4 significant proteins in Bosch et al., however, we were unable to identify any significant proteins in common among the datasets. These are represented in Figures 2A, B volcano plots, showing the upregulated and downregulated proteins among the significant proteins. Proteins found in the univariate analysis were used in the phases of the following analysis, with the 159 from UK Biobank used in the first phase and the 98 of those that were also found in Bosch et al. used in the second phase, since these would be used in the validation tests.
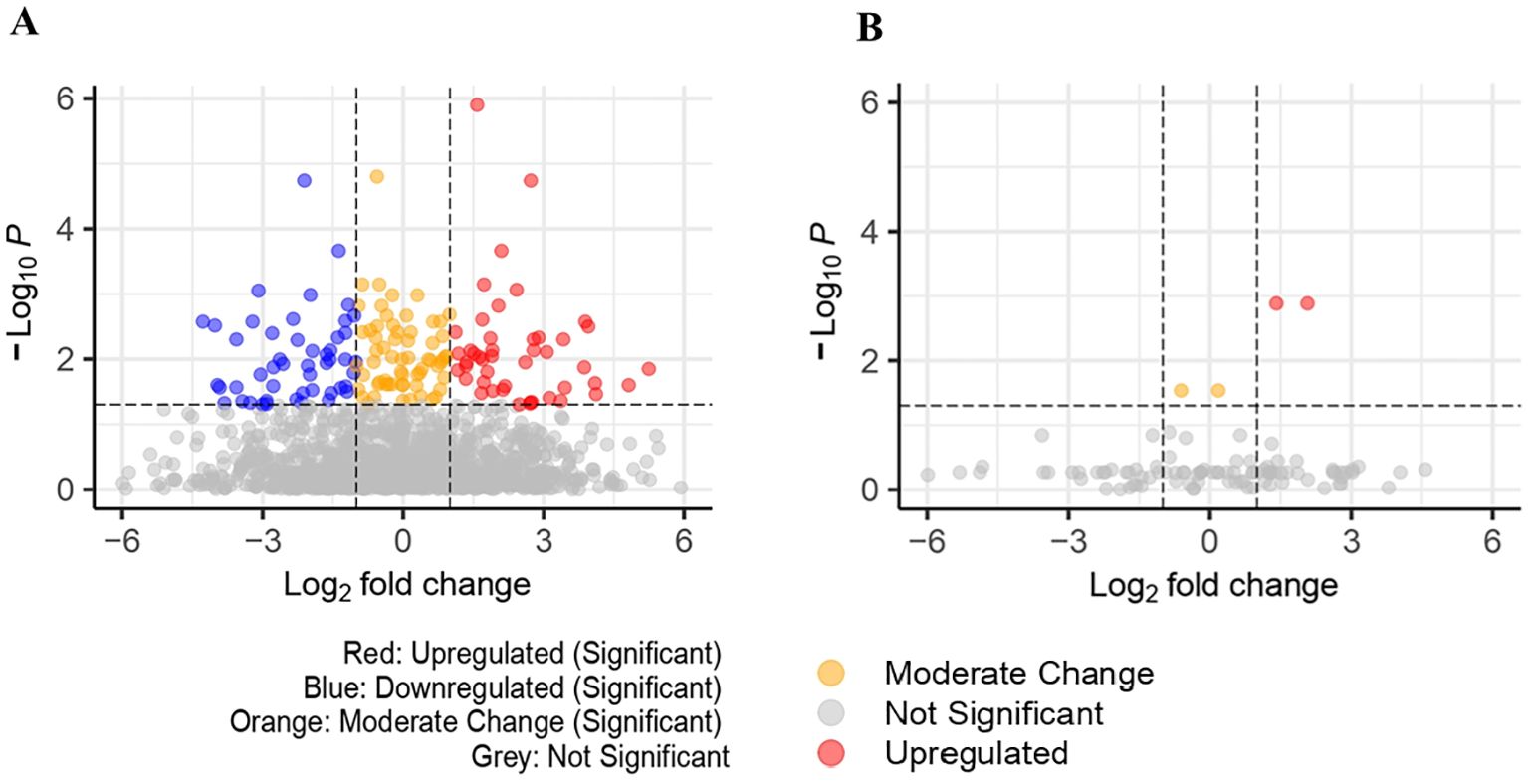
Figure 2. (A) Volcano plot for significant proteins(padj<0.05) in UK Biobank dataset (159 significant proteins). (B) Volcano plot for significant proteins (padj<0.05) in Bosch et al. dataset (4 significant proteins).
3.3 Performance evaluationThe performance of the classifiers was assessed in both phases using eXtreme gradient boosting (XGBoost), light gradient boosting machine (LGBM), and least absolute shrinkage and selection operator (LASSO). Supplementary Tables S2 and S3 summarize the hypertuning parameters used. Supplementary Tables S4 and S5 summarize the performance metrics for each classifier.
With the objective to boosting model accuracy, reducing overfitting, and improving generalization on unseen data, each model’s hyperparameters were defined in specific grids to allow GridSearchCV to test a range of potential values. For XGBoost, the parameters included n_estimators to control boosting rounds, max_depth to set tree complexity, learning_rate to balance learning speed and accuracy, and subsample and colsample_bytree to reduce overfitting through data and feature sampling. Similar parameters were used by LightGBM, but the complexity of each tree was controlled by num_leaves rather than n_estimators. The regularization strength affects feature selection by reducing coefficients towards zero, hence we only defined the alpha parameter for Lasso regression. So as to maximize AUC (Area Under the ROC Curve), a metric that assesses the trade-off between true positive rate (TPR) and false positive rate (FPR) and is especially well-suited to binary classification, we analyzed each model’s performance across these grids and identified the best parameter combinations. GridSearchCV was set up with stratified 5-fold cross-validation using StratifiedKFold for every model. In order to ensure stability and prevent overfitting to any specific data split, this approach splits the data into five parts while preserving class proportions in each fold. GridSearchCV evaluates every parameter combination over all folds using AUC as the evaluation measure. It then chooses the combination that has the greatest average AUC score, hence determining the optimal model configuration. By avoiding overfitting and offering an unbiased evaluation of the model’s performance on the training set, this cross-validation technique enhances the generalizability of the chosen parameters. After determining the optimal parameters, we assessed each model’s performance in the test set. AUC, specificity, sensitivity, precision, and F1-score are among the metrics we calculated to give a thorough evaluation of each model’s strengths and weaknesses. The confusion metrics for each dataset of the two phases in shown in Supplementary Tables S6 and S7.
The classifiers produced quite similar performance metrics for the UK Biobank dataset. LASSO generated the highest AUC test score of 0.75 (95% CI:0.65-0.84), whereas XGBoost scored 0.71 (95% CI:0.61-0.81) and LightGBM 0.70 (95% CI:0.59-0.79). With an optimal grid alpha value for LASSO of 0.01, it was the most effective at detecting individuals with colorectal cancer. LASSO further exhibited some balance between recall and precision with an F1 score of 0.73. The model’s specificity score of 0.67 suggests that it has the potential to accurately detect non-CRC cases. However, the model exhibited more errors in accurately comprehending non-CRC cases (Supplementary Figure S4A). In the second phase, LASSO produced an AUC test score of 0.64 (95% CI:0.52-0.74), again the best of the three classifiers, while XGBoost scored an AUC of 0.63 (95% CI:0.52-0.73) and LightGBM with AUC of 0.60 (95% CI:0.49-0.70). Lasso had an optimal grid value of 0.01 and an F1 score of 0.61. The specificity score of the model of 0.42 indicates that it had little ability to accurately identify instances that were not colorectal cancer (Supplementary Figure S4B).
Performance of the classifiers was markedly worse in the validation dataset (Bosch et.al). When the classifier’s performance was compared, XGBoost and LightGBM performed better with an AUC test of 0.53 (95% CI:0.41-0.66) than LASSO (AUC test = 0.40(95% CI:0.20-0.63). XGBoost and LightGBM detected all positive instances (recall=1) but failed to find any negative predictions (specificity=0) (Supplementary Figure S5A). In phase 2, XGBoost was found to be performing moderately with an AUC test score of 0.61 (95% CI:0.40-0.82) compared to LightGBM AUC test 0.57 (95% CI:0.36-0.77) and LASSO AUC test 0.55 (95% CI:0.35-0.74). The optimal grid parameters of XGBoost were colsample_bytree:0.8, learningrate:0.05, max_depth:5, n_estimators:200, and subsample:0.8. It produced an F1 score of 0.59, which reflects a balance between recall and precision, demonstrating XGBoost was successful in differentiating between colorectal cancer and non-colorectal cancer (Supplementary Figure S5B).
3.4 Feature selection and interpretationAnalysis using SHAP identified 25 proteins (Figure 3A) that were shared among the top 50 proteins in three models in the first phase of UK Biobank data. Of these, two proteins were shared with those found in Bosch et al. After extracting the 98 proteins of UK Biobank in phase 2, 29 of the top 50 proteins of each model were common among three classifiers (Figure 3B). These same 29 proteins were extracted from Bosch et al. and validated using the three models. The 29 common proteins of both datasets were visualized using a heatmap, as seen in Figure 4. One notable difference is that CBLIF and DPEP1 were expressed less in cases found in UK Biobank but higher in Bosch et al. cases.

Figure 3. (A) Common protein (25) among top 50 proteins (3 models) of UK Biobank. (B) Common proteins (29) among top 50 proteins (3 models) of UK Biobank.
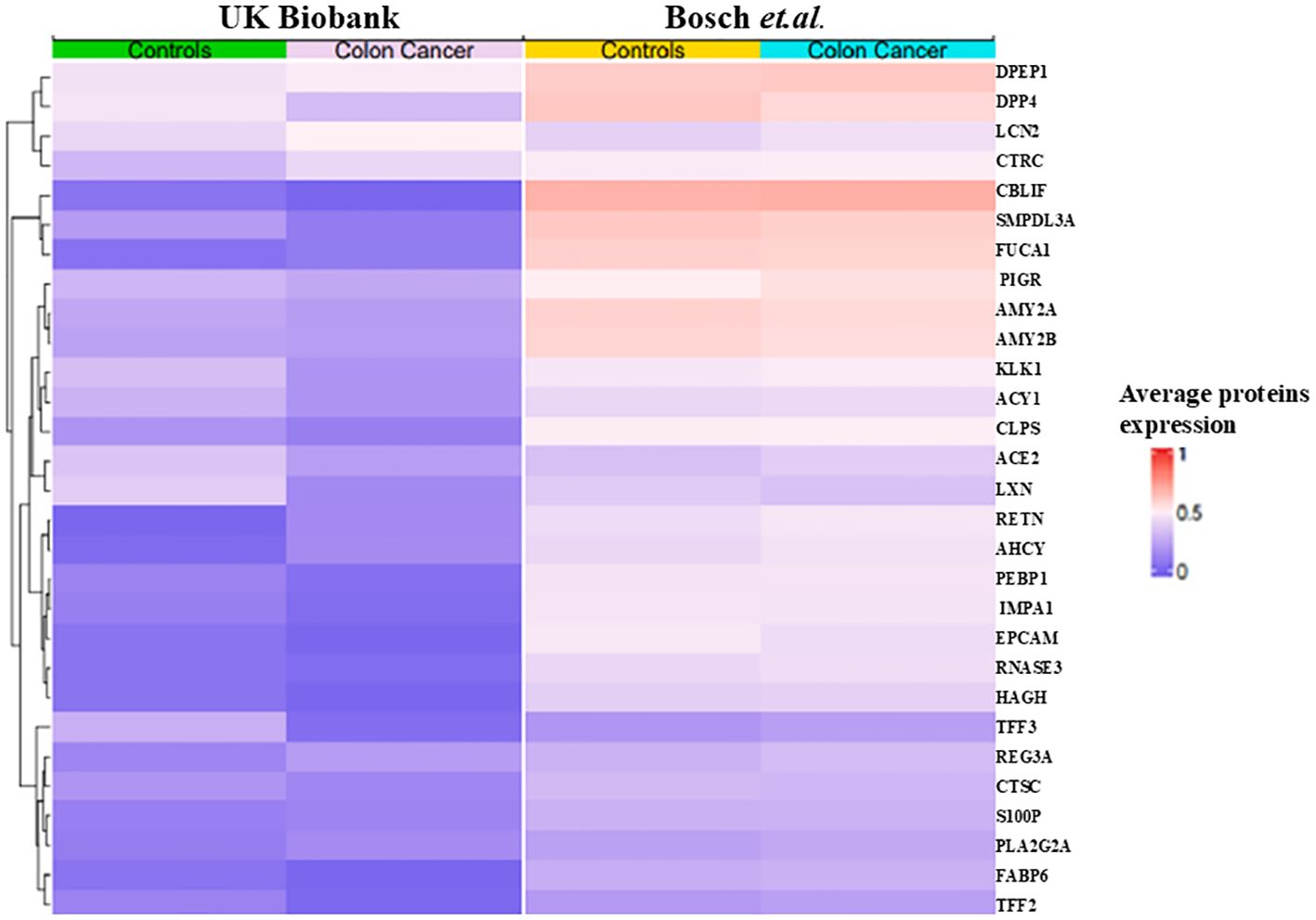
Figure 4. Heat map of 29 proteins of UK Biobank and Bosch et al.
In the first phase, the most important protein was discovered to be the inflammation indicator (63) CEACAM5 in both datasets followed by B4GAT1, MFAP3, and LRRN1 were the next most highly ranked proteins in the UK Biobank data whereas MZB1, and ACE2 were determined to be low ranked proteins. From examining the local model impact plots, CEACAM5, B4GAT1, MFAP3, and LRN1 were observed to influence the model’s prediction of the predictive class CRC. On the other hand, MZB1and ACE2 appeared to be predictive of a lower likelihood in the prediction of colorectal cancer. PLA2G2A did not show up much expression in the Bosch et al. data (Supplementary Figures S6A–D).
The most important proteins found in UK Biobank in phase 2 were discovered to be AHCY and HAGH. Also notable are TMPRSS15 and MEP1B considered less important in Bosch et al. There was higher expression of DPP4 and PLA2G2A in individuals with CRC and the least important protein was TFF2. The proteins AHCY, HAGH, DPP4, and PLA2G2A had elevated levels and were chiefly responsible for the prediction of CRC. TMPRSS15, MEP1B, and TFF2 lowered the prediction of CRC (Figures 5A–D).
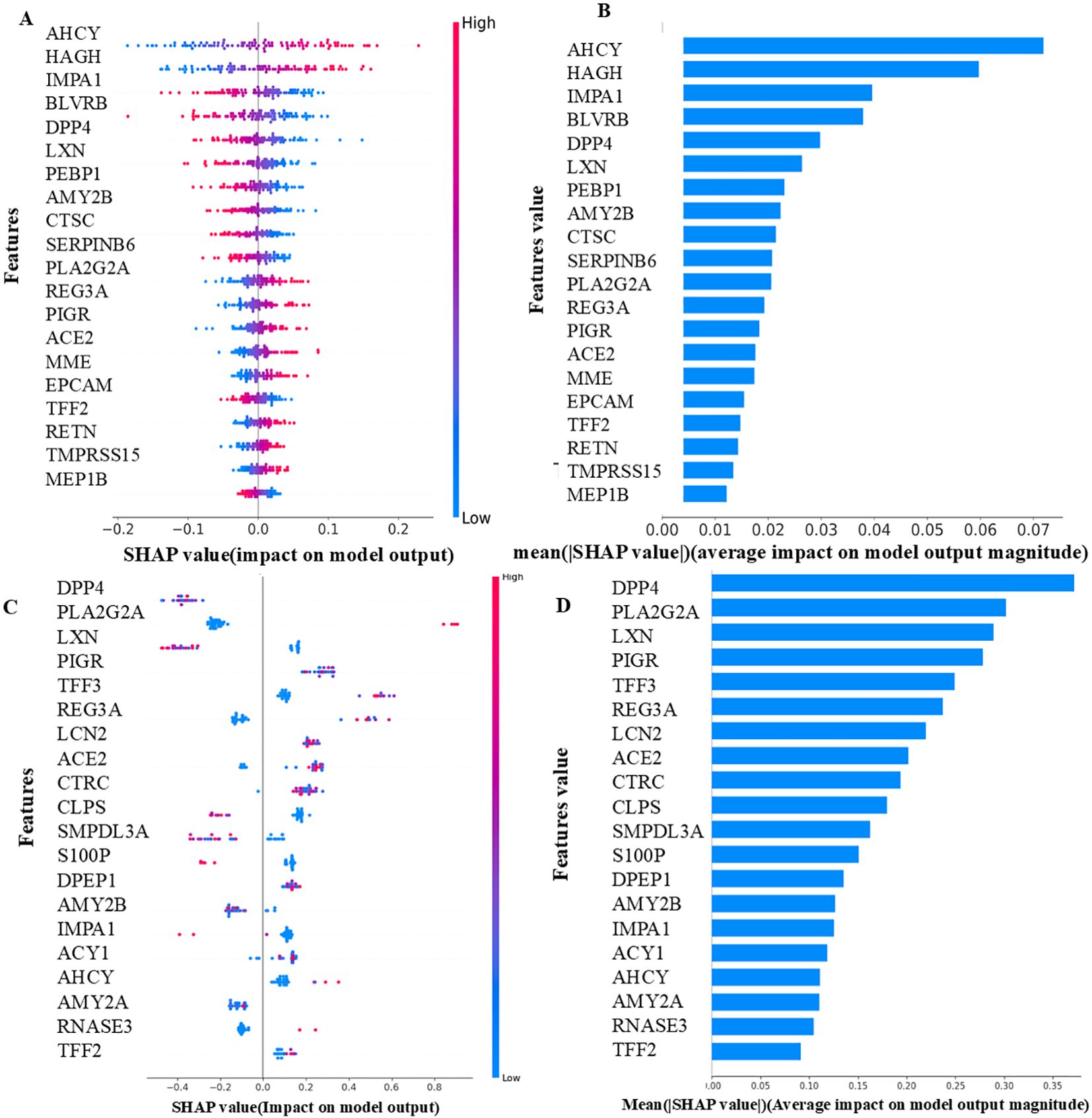
Figure 5. (A) UK Biobank - Local importance (LASSO-Phase 2). (B) UK Biobank - Global importance (LASSO-Phase 2). (C) Bosch et al. - Local importance (XGBoost-Phase 2). (D) Bosch et al. -Global importance (XGBoost-Phase 2). The features displayed are the top 20 proteins of each model, as determined by their SHAP values. The samples are shown as colored dots in the local importance summary plot for SHAP values, the color of each dot corresponds to its value for that feature. Positive SHAP values have a positive effect on the model and direct the algorithm to predict the positive class, and vice versa. In the global importance summary plot for mean absolute SHAP values higher rank features are associated with more samples having SHAP values.
3.5 Interaction networks using STRING and the Human Protein AtlasThe proteins common in the two datasets were given as input into the STRING database to identify interactions. The protein CD163, one of the 25 proteins (Supplementary Figure S7A) identified in the first phase, was shown to have a strong correlation with other proteins, including IL2RA, HGF, LAG3, SELE, and to be crucial for both cell adhesion and metastasis. The inflammatory protein CEACAM5 had a strong correlation with TFF1 and LAG3 proteins. Analysis of 29 proteins (Supplementary Figure S7B) in phase 2 revealed that AMY2A and AMY2B had evidence of a strong interaction in pancreatic cancer, while our study did not find a significant correlation between them. Experimentally, it was discovered that TFF3 and TFF2 were highly correlated.
Seven proteins, TFF3, LCN2, CEACAM5, TFF1, SELE, RETN, and AHCY were found to be upregulated in other databases and also in our UK Biobank where TFF3 and LCN2 were found to be the most significant proteins and were highly expressed in colorectal cancer. The comparison of seven proteins in the case and control groups is illustrated (Supplementary Figures S8A–G).
The prediction of both dataset models was tested with SHAP XAI, which also highlighted the key proteins influencing the prediction of the models. The top proteins found in the UK Biobank included CEACAM5, B4GAT1, and AHCY. However, the model’s average AUC score of 0.69 implies that these proteins are not effective in classifying colorectal cancer samples in UK Biobank and cannot distinguish between CRC cases and controls. However, given that the results are not as low as chance 0.50, it is acceptable to make the assumption that the pathophysiology of CRC is influenced by these important proteins, with findings supported by the literature mentioned below. According to previous studies CEACAM5 (64), B4GAT1 (65), and AHCY (66) were discovered to be biomarkers of CRC due to their strong involvement in the methylation and inflammatory processes of tumor cells. These were discovered to be inverse biomarkers that correspond with the CRC prediction made by our UK Biobank models.
Seven proteins namely TFF3, LCN2, CEACAM5, TFF1, SELE, RETN, and AHCY proteins of both phases were found to be upregulated in colorectal cancer in human protein atlas database, also were significant in our UK Biobank dataset and had an essential function in CRC (Supplementary Table S8).
TFF3 and LCN2 were found to be the top most significant proteins in the UK Biobank.TFF3 and LCN2 were found to be highly expressed in other cancers like Myeloma, and lung cancer in the human protein atlas database.TFF3 was the most significant protein found in our study and played an important role in the proliferation, migration, and invasiveness of HT29 cells in colorectal cancer (67).TFF3 has a physical association with PCBD1, UBQLN2, and SGTA which were considered to be transcription factors and other protein classes in the human protein atlas database (Supplementary Figure S9A). TFF3 plays an important role in the apoptosis, and cell proliferation along with the promotion of angiogenesis in colorectal cancer (68) (Supplementary Figure S9B). It interacts with tyrosine kinase (src) protein and activates signal transducer and activator of transcription 3 (STAT3) which is plays an important role in the signaling pathway of cancer progression (69). A genome-wide association study (GWAS) analysis revealed that chromosome 21 is known to have the TFF3 protein. The expression of TFF3 was found to be high in our CRC samples.
LCN2 appeared to be the 2nd most significant protein and played an important role in the enzymatic activity of matrix metalloprotease-9 causing metastasis of colorectal cancer cells (70) It has a physical association with several other transcription factors and other protein classes (Supplementary Figure S10A). They played an important role in tumor cell growth, iron toxicity, and methylation and served against the anti-cancer drugs (71) (Supplementary Figure S10B). GWAS analysis revealed that chromosome 9 is known to have the LCN2 protein.
3.6 Weighted gene co-expression network analysis resultsIn our assessment of the UK Biobank dataset, we evaluated the scale-free topology model fit and mean connectivity to determine its suitability for WGCNA. Although the mean connectivity was not atypical, the consistently low signed R² values indicated a poor fit to the scale-free topology model (Supplementary Figures S12A, B). This suggested that the dataset did not fully exhibit the scale-free network characteristics typically required for robust co-expression analysis. As a result, the dataset was deemed unsuitable for further WGCNA analysis and no modules were identified or additional analyses, such as module-trait correlations or functional enrichment were performed.
In the Bosch et al. dataset the chosen β value of 8 was supported by a scale-free topology model fit (signed R²) consistently achieving a value of 0.90 across tested β values, indicating a robust scale-free network (Figure 6A). The mean connectivity plot showed a decline with increasing β, suggesting a sparser network structure, with the selected β ensuring a balance between scale-free topology and network interpretability (Figure 6B).
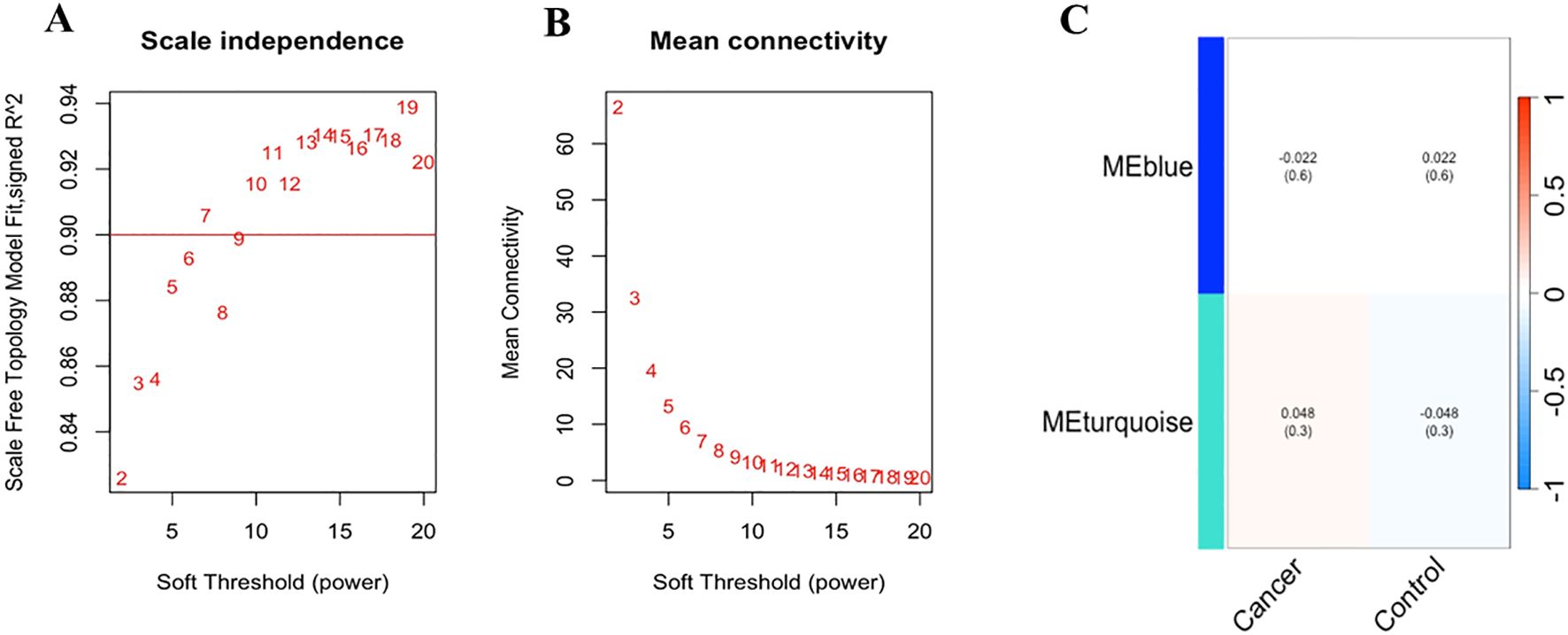
Figure 6. (A) Scale-free topology model fit and mean connectivity analysis of Bosch et.al., dataset: The horizontal line at a signed R² value of 0.90 indicates that the network exhibits a good scale-free topology fit across the tested range of soft-thresholding power (β) values, suggesting that the network’s degree distribution closely follows a power-law distribution. (B) The mean connectivity for different β values shows as β increases, the mean connectivity decreases, indicating a reduction in the number of connections per node, which is consistent with a sparser network structure. The selected optimal β value of 8 balances. (C) The heatmap illustrates the correlation between the identified module eigengenes (MEs) and the defined traits, “Cancer” and “Control” (non-cancer). The rows represent the MEs (MEblue and MEturquoise), and the columns represent the traits. Each cell contains the Pearson correlation coefficient, with the corresponding p-value provided in parentheses.
Subsequently, module detection via hierarchical clustering on the Bosch et al. dataset identified two distinct modules, MEBlue and METurquoise, with the remaining genes assigned to the Gray module, indicating no significant co-expression. The cluster dendrogram visually confirmed these modules, with MEBlue and METurquoise displayed prominently (Supplementary Figure S11).
To explore the functional relevance of the identified modules, we correlated the module eigengenes (MEs) with the defined clinical traits (Figure 6C). The MEBlue module exhibited a low negative correlation with cancer (r = -0.022, p = 0.6) and a low positive correlation with control (r = 0.022, p = 0.6), suggesting no significant differential expression in relation to these traits. In contrast, the METurquoise module showed a slightly positive correlation with cancer (0.048, p = 0.3) and a slightly negative correlation with control (r = -0.048, p = 0.3), indicating a marginal trend of up-regulation in cancer samples.
Further functional annotation through KEGG pathway enrichment analysis revealed that genes in the METurquoise module were significantly associated with several biological pathways, with the MAPK signaling pathway being the most enriched. This suggests a potential involvement of these genes in critical cellular processes such as proliferation and survival, which is relevant to cancer pathology.
Overall, our WGCNA analysis has identified key gene modules and potential functional pathways associated with cancer. While the correlations with clinical traits were modest, the identification of the signaling pathways highlighting potential biological mechanisms calls for further investigation. This experiment provides a comprehensive framework for understanding gene co-expression patterns and their potential functional implications in colorectal cancer.
3.7 Pathway-based interpretationFigure 7 explains how each of the biomolecules is capable of triggering colorectal cancer through various proteogenomic pathways. Trefoil factor 3 (TFF3) has been found to promote cell migration and increase proliferation of colorectal cancer cells. Overexpression of TFF3 in colorectal cancer cells decreases the levels of E-cadherin which results in increased epithelial-mesenchymal transition, enhancing colon cell migration and promoting the formation of secondary tumors, thereby progressing cancer (67). Decreased levels of E-cadherin activate the EGF receptor signaling cascade leading to phosphorylation of β-catenin and activation, altering cell-cell interactions and leading to cell migration (72). TFF3 has also been found to activate signaling pathways that promote cellular invasion including src/RhoA, PI3K/Akt, and phospholipase C (PLC)/Protein kinase C (PKC) pathways. By activating the PI3K/Akt signaling pathway, cell survival and invasion are enhanced (73). Activation of this pathway also leads to downstream effects such as inhibition of pro-apoptotic factors and activation of proteins that promote protein synthesis and cell growth. The PLC/PKC pathway is also involved in cellular motility and invasion, which is an important step in the metastatic spread of cancer cells (73). Compared to healthy cells, in colorectal cancer cells levels of Lipocalin 2 (LCN2) are elevated (74). In colorectal cancer cell lines LCN2 overexpression was linked to increased invasion of cells and loss of cell-to-cell adhesion (74). LCN2 has been shown to protect matrix metalloproteinase 9 (MMP9) from proteolytic degradation by forming an LCN2/MMP9 complex (74). MMP9 plays an important role in the resorption of the extracellular matrix and therefore in metastasis and neoplastic invasion (75). By covalently bonding to MMP9, LCN2 can decrease the degradation of MMP9 and therefore increase tumor progression by enhancing the enzymatic activity of MMP9 (75, 76). Carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM-5) is a glycoprotein overexpressed in colorectal cancer (77). CEACAM-5 inhibits anoikis, a type of apoptosis that is triggered by the loss of extracellular matrix-cell contacts, therefore disrupting colonic tissue architecture (77, 78). CEACAM5 interacts with DR5, a member of the death receptor family found on the plasma membrane of colorectal cancer cells that have detached from the extracellular matrix. This leads to reduced caspase-8 activation therefore leading to the inhibition of caspase-3 (79). CEACAM-5 is clustered in lipid rafts and activates integrin-α5 which activates the PI3k/Akt signaling pathway which promotes cell survival. Downstream effects of activating the PI3K/Akt signaling pathway include inhibition of the release of cytochrome-c from the mitochondria, resulting in the prevention of apoptosis in detached cells and increasing migration of the cell allowing the formation of a secondary tumor (78).
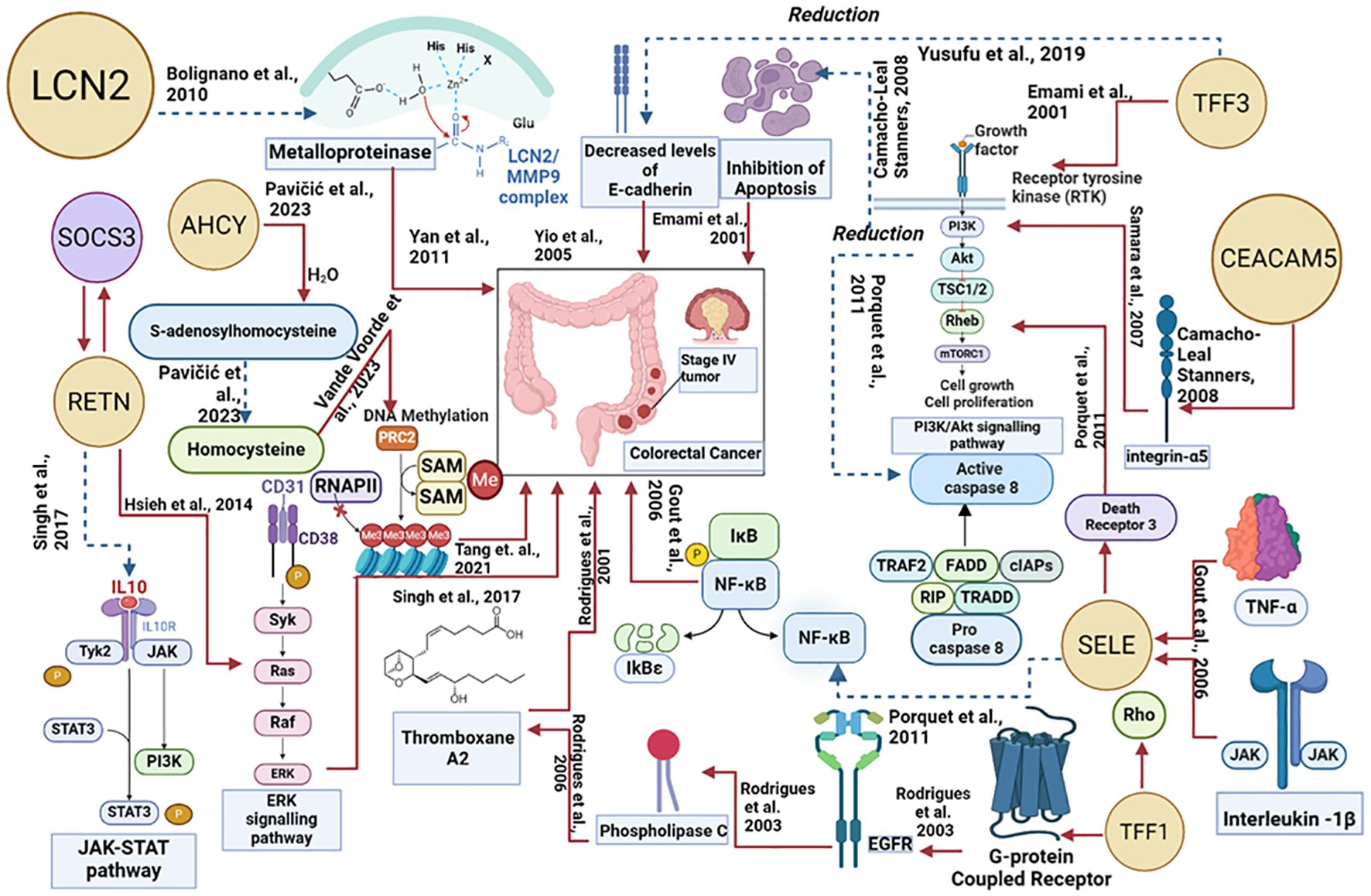
Figure 7. Proteogenomic insights into the biological pathways of proteins associated with colorectal cancer. The red line denotes the upregulated actions, and the blue dotted lines denote the downregulated actions.
Trefoil factor 1 (TFF1) overexpression facilitates tumor growth and invasiveness through various pathways including Rho-GTPases, Rho-kinase, PI3-kinase, PLC, and COX-2 (73, 80).TFF1-induced cellular invasion is dependent on the EGFR signaling pathway.TFF1 indirectly activates EGFR, through a mechanism involving the transactivation of EGFR using G-protein coupled receptors (GPCRs) (73)TFF1 can also increase invasiveness through Thromboxane A2 (TXA2) receptor/PLC - dependent mechanisms. TFF1 upregulates the production of TXA2 which binds to the TXA2 receptor which is coupled to G-proteins and goes on to activate PKC. PKC enhances cell proliferation, survival, and invasion therefore progressing colorectal cancer (73). Selectin-E (SELE) levels are significantly higher in colorectal cancer cells in comparison to adjacent healthy cells (55). SELE is induced by pro-inflammatory stimuli such as tumor necrosis factor-α and IL-1β (81, 82). SELE is able to bind death receptor 3 (DR3) and activate it leading to the recruitment of src kinase. Src kinase phosphorylates the tyrosine residues on DR3 leading to activation of PI3K. PI3K activates Akt leading to a variety of downstream effects including the activation of the NFκB pathway. The PI3K/Akt/NFκB pathway is known to protect colorectal cancer cells from apoptosis by reducing the activity of caspase-8 and caspase-3. These caspases are important in the induction of apoptosis, suggesting that SELE increases the survival of cells and resistance towards apoptosis, further progressing the tumor (82). Resistin (RETN) is overexpressed in colorectal cancer (83). RETN binds to toll-like receptor 4 (TLR4) on the surface of colorectal cancer cells, switching on ERK signaling (83, 84). Activated ERK can promote upregulation of the gene SOCS3 which leads to decreased phosphorylation of JAK2 and STAT3 (83). Inhibition of the JAK2/STAT3 signaling pathway results in the growth arrest of cells in the G1 phase of the cell cycle, therefore regulating cell growth (83). S-adenosylhomocysteine hydrolase (AHCY) levels in colorectal cancer are disrupted, leading to an imbalance in methylation processes. AHCY is an enzyme that catalyzes the hydrolysis of S-adenosylhomocysteine (SAH) into homocysteine. Increased activity of AHCY leads to increased conversion of SAH into homocysteine. SAH inhibits the activity of methyltransferases and as AHCY inhibits the accumulation of SAH, there is more DNA methylation occurring leading to abnormal gene expression which contributes to tumor progression (66).
3.8 Protein quantitative trait loci analyses resultsSignificantly associated loci were identified in the groups with prior CRC and that would go on to be diagnosed with CRC after sampling. In those who had been previously diagnosed with CRC, this included 57 SNPs associations with SELE levels and for those who went on to be diagnosed, there were 67 SNPs associated with CEACAM5, 136 with SELE, and 1 with TFF1. However, all of these points were more significantly associated by p-value in the control group and in SNPs whereby significant association was found in a CRC cohort, the Beta coefficient always had the same sign as in the control cohort, indicating the SNP significantly associated had a similar effect to the control group (Supplementary Figure S13, Supplementary Table S9).
3.9 Impact of batch-effect correction on model outcomesA harmonized dataset of 98 common proteins from the UK Biobank and Bosch et al. databases were used to evaluate the performance of prediction models such as XGBoost, LightGBM, and LASSO regression. AUC was 0.60, specificity was 0.58, recall was 0.57, accuracy was 0.59, and F1 was 0.58 for LASSO regression. The AUC score for both XGBoost and LightGBM was 0.57. XGBoost’s specificity was 0.52, sensitivity was 0.62, precision was 0.58, and F1 score was 0.60. LightGBM’s specificity was 0.49, recall was 0.66, precision was 0.57, and F1 score was 0.61.
LightGBM obtained an AUC score of 0.62, specificity of 0.52, recall of 0.66, accuracy of 0.59, and F1 score of 0.62 for the seven potential biomarkers. An AUC score of 0.60, specificity of 0.49, recall of 0.62, accuracy of 0.56, and F1 score of 0.59 were all attained with LASSO regression. With an F1 score of 0.59, recall of 0.60, accuracy of 0.57, specificity of 0.52, and AUC of 0.57, XGBoost performed well. The proteins AHCY, LCN2, CEACAM5, TFF3, TFF1, SELE, and RETN were shown to be the most significant when the highest AUC model LightGBM model was subjected to SHAP analysis to ascertain feature importance, suggesting their possible applicability in predictive modeling. The distribution of the seven proteins of the ComBat-corrected dataset is depicted (Supplementary Figure S14).
3.9.1 Comparative validation of potential biomarkers across CRC studiesThe expression levels of TFF3 and TFF1 in colorectal cancer tissues were examined in a study conducted by Yusufu et al. (67) which revealed TFF3 and TFF1 expression levels are elevated in colorectal cancer and promote the malignant behavior of colon cancer by activating the EMT process. The mRNA expression levels of these proteins were assessed by the researchers using quantitative real-time P
留言 (0)