
記住我
Graves disease (GD) is an autoimmune disease that affects the thyroid gland resulting in hyperthyroidisms and goiter (enlargement of the thyroid gland); therefore, GD is also known as a toxic diffuse goiter (Davies et al., 2020). The name of GD is derived from the Irish doctor Robert James Graves who described a case of GD in 1835. Subsequently, in 1840, the German physician Karl Adolph von Basedow independently described the same features of GD (Weetman, 2003), thus leading to the alternate name of Basedow disease (Leporati et al., 2015) among others. The basic clinical feature of GD includes the symptoms of hyperthyroidism such as weight loss, sweating, tachycardia, and diarrhea (Leporati et al., 2015). Approximately 25%–30% of patients with GD have extra-thyroidal manifestations such as exophthalmos (eye bulging) or Graves opthalmopathy; accordingly, GD is also known as exophthalmic goiter (Cao et al., 2022). The pathognomonic features of GD are hyperthyroidism, goiter, exophthalmos, and pretibial myxedema that not present in other types of hyperthyroidism (Manni et al., 2020). The thyroid-associated opthalmopathy (TAO) develops in GD due to stimulation of thyroid stimulating hormone receptor (TSHR) of fibroblasts and hypertrophy of muscles around the eyes (Neag and Smith, 2022).
The incidence of GD is about 7.5 times more common in women than men. It is more frequent in the age of 40–60 years, although it can happen at any age (Hussain et al., 2017). GD is regarded as the most common cause of hyperthyroidism in the United States (Bartalena, 2013). In addition, GD is often associated with other autoimmune diseases such as rheumatoid arthritis and type 1 diabetes suggesting the immune disorders of GD (Ferrari et al., 2019; Al-Kuraishy et al., 2024b). Of note, genetic causes are also involved in the pathogenesis of GD (Brix et al., 2001). For example, HLA-DRB3/DR3 (major histocompatibility complex, class II, DR beta 3) increases the susceptibility for the induction of GD (Inaba et al., 2016). As well, single gene defects are also linked with the induction of GD. For example, PTPN22 (protein tyrosine phosphatase non-receptor type 22) and CTLA4 (cytotoxic T-lymphocyte associated protein 4) gene mutations are implicated in the pathogenesis of GD (Pujol-Borrell et al., 2015). Moreover, the autoimmunity of GD is triggered by viral and bacterial infections due to antigenic mimicry. For example, Yersinia enterocolitica has a structural similarity with human TSH receptor and infection by this organism results in the induction of GD (Hargreaves et al., 2013). Likewise; Epstein-Barr virus is considered as a potential trigger of GD (Pyzik et al., 2019).
Although the main cause of GD is not clearly defined, the primary proposed mechanism involves autoantibodies that activate TSHRs; hence, these autoantibodies are known as TSIs (thyroid-stimulating immunoglobulins) (Mathew et al., 2021). TSIs activate TSHRs of the thyroid gland resulting in excessive release of thyroid hormones with subsequent development of hyperthyroidism and goiter (Mathew et al., 2021).
The underlying mechanisms for the development of autoimmunity in GD are related to the autoactivation of T and B cells with subsequent generation of autoantibodies against TSHRs (Liu et al., 2018). Of note, both cellular and humoral immunity are intricately involved in the pathogenesis of GD. Type 1 T helper (Th1) and Th2 cells are highly involved in the induction of abnormal immune response and the pathogenesis of GD (Antonelli et al., 2020). Th1 through activation of cytotoxic lymphocytes and macrophages affect the proliferation of thyroid follicular cells (Pierman et al., 2021). However, Th2 triggers the production and activation of B lymphocytes and plasma cells resulting in the generation of TSIs against TSHRs (He et al., 2020). In GD, the stimulatory activity of TSI is mainly present in the IgG1 subclass, which is chiefly activated by Th1 cells (Li et al., 2021d). As well, Th1 promotes the generation of TSIs from B lymphocyte via an IL10-dependent pathway (Pierman et al., 2021). Moreover, exaggerated Th17 also activates abnormal TSI in GD (Torimoto et al., 2022). However, regulatory T/Treg cells, which downregulate the abnormal immune response are highly reduced leading to the induction of an abnormal immune response in GD (Fang et al., 2021). In addition, in GD the B lymphocytes are autoactivated due to the downregulation of regulatory B/Breg lymphocytes, also leading to an abnormal immune response and activating the release of TSIs (Wang et al., 2021). Therefore, an abnormal immune response and the development of autoimmunity are involved in the pathogenesis of GD (Figure 1).
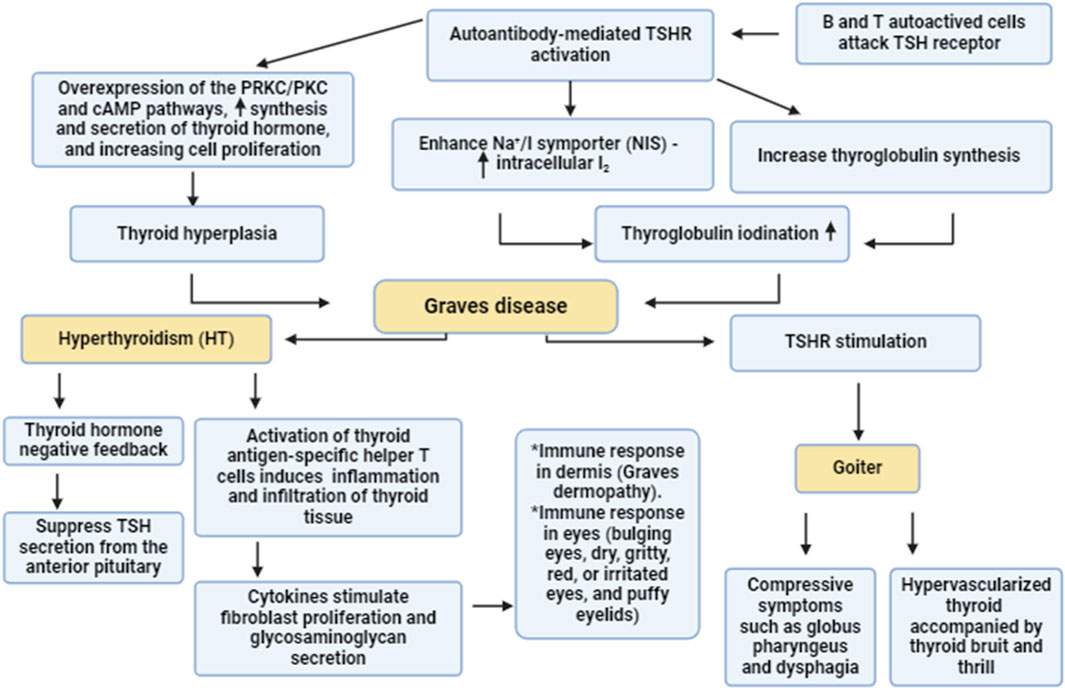
Figure 1. Pathogenesis of GD. Autoreactive B and T cells induce the formation of autoantibodies which activate thyroid stimulating hormone receptors (TSHR). Activated TSHR activates the synthesis and release of thyroid hormones via PKC/cAMP which promote thyroid hyperplasia. In addition, activated TSHR stimulates thyroglobuline synthesis and enhances intracellular accumulation of I2 through activation of Na+/I symporter. These changes trigger the development of GD which causes goiter and hyperthyroidism.
Different studies highlight a potential crosstalk between autophagy and the immune response, and the former plays a critical role in autoimmunity (Wu and Adamopoulos, 2017). Autophagy is intricately involved in the expression of intracellular genes, the initial immune response, and cytokine release (Bhattacharya and Eissa, 2013). Autophagy inhibition ameliorates many autoimmune diseases such systemic lupus erythematosus/SLE and rheumatoid arthritis. Conversely, autophagy inhibition exacerbates Crohn disease and psoriasis (Bhattacharya and Eissa, 2013; Wu and Adamopoulos, 2017). It has been reported that autophagy is implicated in the pathogenesis of GD and other thyroid diseases through the modulation of immunity and the inflammatory response (Duan et al., 2019; Chen et al., 2023). In addition, autophagy is also implicated in the pathogenesis of TAO (Duan et al., 2019; Chen et al., 2023). However, the exact role of autophagy in GD is not well explained. Therefore, this review discusses how autophagy plays an integral role in the pathogenesis of GD regarding its protective and harmful effects.
Autophagy and molecular signalingAutophagy is an evolutionarily conserved cellular process that promotes the survival of eukaryotic cells in response to different exogenous and endogenous stimuli such as starvation (Ali et al., 2023; Marzoog et al., 2023; Al-Kuraishy et al., 2024c; Sulaiman et al., 2024). Autophagy is involved in the elimination of damaged or superfluous organelles and proteins, recycling the breakdown products that result from their degradation for cellular nutrition (Li et al., 2021b; Al-Kuraishy et al., 2024a). Autophagy plays critical roles in different biological functions in normal and disease states. This process is crucial in the regulation of inflammation, immunity, stress adaptation, cancer, aging and neurodegenerative diseases (Luo et al., 2020). A key feature of autophagy is that degradation occurs through the lysosomal pathway (Cao et al., 2021). There are various types of autophagy that differ in terms of the mechanism as well as substrates (Galluzzi et al., 2017). The predominant form of autophagy, macroautophagy (hereafter autophagy) is initiated by the formation of a phagophore which is sequesters cytoplasmic components and then matures into a double-membrane autophagosome (Broggi et al., 2020). The autophagosome fuses with an endosome and/or a lysosome to form an autolysosome where the contents are degraded; the resulting macromolecules are then released back into the cytosol (Broggi et al., 2020) (Figure 2).
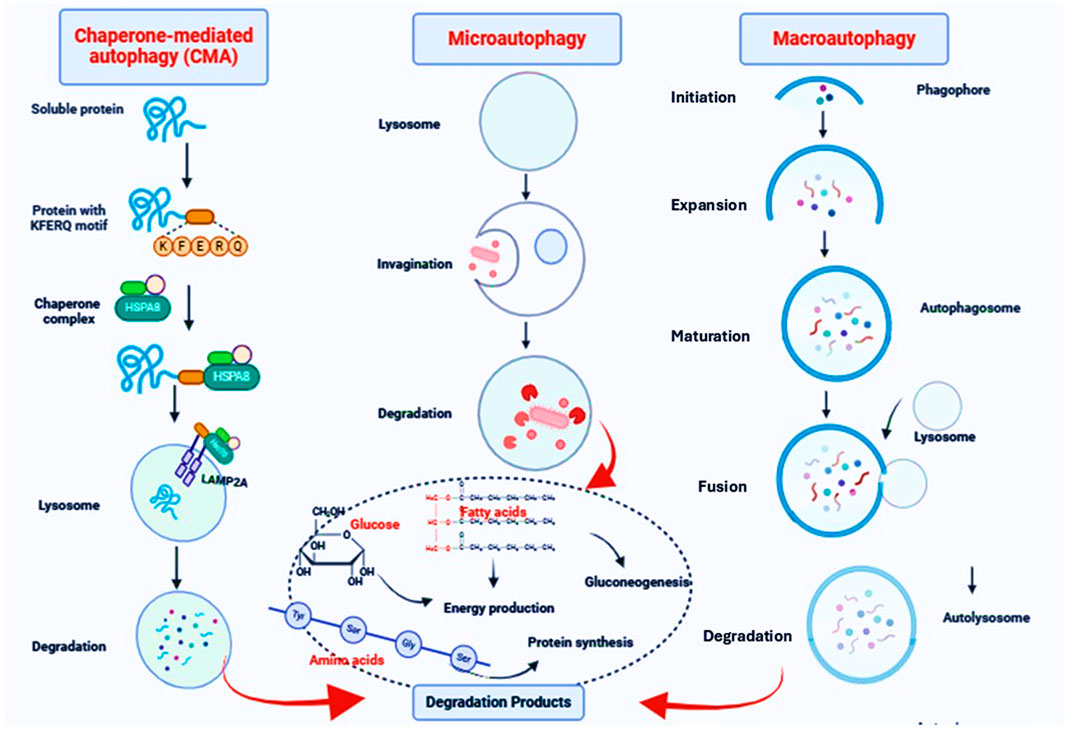
Figure 2. The autophagy pathway. Macroautophagy is initiated by the formation of a phagophore which is sequesters cytoplasmic components and then matures into a double-membrane autophagosome. The autophagosome fuses with an endosome and/or a lysosome to form an autolysosome where the contents are degraded; the resulting macromolecules are then released back into the cytosol. In microautophagy, the limiting membrane protrudes or invaginates at the surface of the lysosome/vacuole by the lateral segregation of lipids and local exclusion of large transmembrane proteins, which is conducted at the small smooth areas with a very low content of transmembrane proteins. In CMA, proteins, the only cargo degraded by this pathway, cross the lysosomal membrane one by one. Not all proteins can undergo degradation via CMA. To be CMA substrates, proteins must contain a specific targeting motif in their amino acid sequence. This motif binds to a cytosolic chaperone (HSPA8), which brings the unfolded substrate protein to the lysosomal surface for internalization and rapid intralysosomal degradation.
Autophagy is coordinated by ATG (autophagy related) proteins. Phagophore formation is initiated by ULK1 (unc-51 like autophagy activating kinase 1)/ULK2 which forms a complex with ATG13, RB1CC1 and ATG101 (Li et al., 2020). This step also requires the class III phosphatidylinositol 3-kinase complex that includes PIK3C3/VPS34, PIK3R4/VPS15, BECN1, ATG14 and NRBF2 (Pavlinov et al., 2020). In addition, BECN1 interacts with other binding proteins such as UVRAG (UV radiation resistance associated), AMBRA1 (autophagy and beclin 1 regulator 1) and SH3GLB1/BIF-1 (SH3 domain containing GRB2 like, endophilin B1) which form various class III complexes (Chang and Zou, 2020). Two ubiquitin-like conjugation systems involving the ATG12–ATG5-ATG16L1 complex, and MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) along with the ATG2-ATG9 lipid transferase-scramblase complex are essential for the expansion of the phagophore (Iriondo et al., 2023); the conversion of LC3-I to the LC3-II lipidated form reflects autophagosome formation (Ueno and Komatsu, 2020). Fusion of autophagosomes with lysosomes requires different components such as UVRAG, VPS proteins and RAB7 (Lőrincz and Juhász, 2020). Autophagy is highly regulated to maintain optimal levels-either too little or too much can be deleterious to cellular physiology. For example, stress and nutrient depletion activate adenosine monophosphate-activated protein kinase/AMPK and inhibit MTOR (mechanistic target of rapamycin kinase) resulting in the activation of the ULK1 and phosphatidylinositol 3-kinase complexes (He, 2022) (Figure 3).
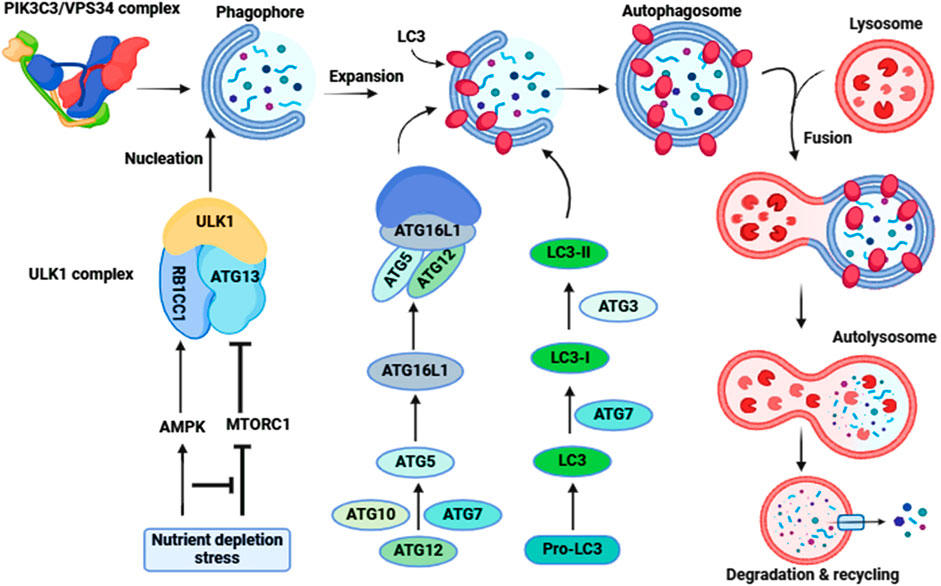
Figure 3. Molecular signaling of autophagy. The ULK1-ATG13-RB1CC1-ATG101 complex is activated to induce autophagy (Initiation). Following the induction of autophagy, an omegasome is formed from the ER by association with ZFYVE1/DFCP1. Next is the formation of a phagophore, which expands to engulf cytoplasmic components, including mitochondria and endoplasmic reticulum (Expansion). Association with the ATG12–ATG5-ATG16L1 complex forms the phagophore. LC3-II localizes to the phagophore membrane at the latter step of autophagosome formation, while the ATG12–ATG5-ATG16L1 complex dissociates from it. Finally, the phagophore membrane is enclosed to form an autophagosome (Maturation). After autophagosome formation, the lysosome fuses with the autophagosome (autophagosome-lysosome Fusion) to form an autolysosome. Intra-autophagosomal contents are degraded by lysosomal hydrolases (Degradation). After formation of the autolysosome, the lysosomal hydrolases degrade the intra-autophagosomal contents, including LC3-II.
Role of autophagy in GDOf note, thyrocyte basal autophagy is essential for the survival of thyroid follicular cells (Kurashige et al., 2019). Kurashige et al. (2019), found that atg5 gene knockout mice experience abnormal morphology and function of thyrocytes with progressive apoptosis. As well, an imbalance of autophagy and apoptosis triggers the development of thyroid damage in rats (Li et al., 2021c). A reduction of autophagy and augmentation of apoptosis are observed in patients with hyperthyroidism due to excess iodine intake (Xu et al., 2016), and induction of the development and progression of autoimmune thyroid disease (AITD) in animal models is mediated by inhibition of autophagy (Duan et al., 2019). Di-isononyl phthalate/DINP-induced AITD occurs through inhibition of normal autophagy via an MTOR-AKT-dependent pathway. Supporting this finding, inhibition of the MTOR pathway by rapamycin attenuates the development of AITD (Duan et al., 2019). GD is regarded as one of most common AITDs and its pathogenesis is highly affected by the MTOR-AKT pathway (Li et al., 2015). In a GD mouse model, the MTOR-AKT pathway is exaggerated and correlates with signs of hyperthyroidism. Treatment in this model with the antithyroid medication methimazole reduces activity of the MTOR-AKT pathway and mitigates GD pathology (Li et al., 2015; Al-Kuraishy et al., 2021; AlAnazi et al., 2023). Zhang et al. (2023) observed that rapamycin mitigates TAO in patients with GD by inhibiting cytotoxic T lymphocytes, which have an upregulated MTOR pathway. Importantly, isolated IgG from GD patients induces the chemoattractant activity of cytotoxic T lymphocytes via the MTOR pathway (Zhang et al., 2023).
Of note, stimulatory TSIs induce survival and proliferation of thyrocytes, whereas blocking TSIs leads to the inactivation of thyrocytes. Moreover, neutral TSIs, which induce apoptosis of thyrocytes can activate the MTOR pathway and cause a downstream decrease in the proliferation of thyrocytes (Morshed et al., 2010). These findings suggest that activation of the MTOR pathway in AITDs such as GD may be responsible for the inhibition of thyroid autophagy and the progression in the pathogenesis of GD. Thus, restoration of thyroid autophagy may reduce the pathogenesis of GD. Indeed, findings from preclinical studies illustrate that the addition of TSH or the antioxidant N-acetyl-L-cysteine/NAC to rat thyroid FRTL-5 cells activates autophagy and attenuates apoptosis (Morshed et al., 2022). The evidence for autophagy activation is shown by an increase in the levels of SQSTM1/p62, ULK1, LC3A, LC3B and BECN1 as well as PRKN- and PINK-related proteins (Morshed et al., 2022). Therefore, enhancement of thyrocyte autophagy prevents TSI-induced apoptosis in GD. Conversely, Faustino et al. (Faustino et al., 2018) showed that IFNA/IFN-α (interferon alpha) induces AITD through induction of autophagy and lysosomal-dependent degradation of TG (thyroglobulin) in human thyroid cells. Moreover, defective thyrocyte autophagy induces apoptosis of thyroid follicular cells by activating the generation of reactive oxygen species/ROS in patients with Hashimoto disease (Lu et al., 2018). These findings indicate that thyroid autophagy is dysregulated in AITDs including GD.
The physiological level of TSH activates thyrocyte autophagy in PCCL3 cells via cAMP-PRKA/PKA as evidenced by increasing levels of LC3 and SQSTM1; however, thyroid hormones T4 and T3 inhibit thyrocyte autophagy (Kurashige et al., 2020). Moreover, many studies confirmed that TSH activates autophagy in muscles, liver and adipose tissue (Sinha et al., 2015; Lesmana et al., 2016; Yau et al., 2019). However, Xin et al. (2017) illustrated that TSH inhibits autophagy in chondrocytes. Therefore, the effect of TSH on thyrocyte autophagy is difficult to interpret as TSH through activation of PRKA promotes the activation of MAPK/ERK, CREB and MTOR which differentially affect autophagy activation (Figure 4). These findings suggest that the action of TSH and TSI differs downstream as TSI promotes the MTOR pathway whereas TSH mainly activates MAPK/ERK and CREB (Kurashige et al., 2020).
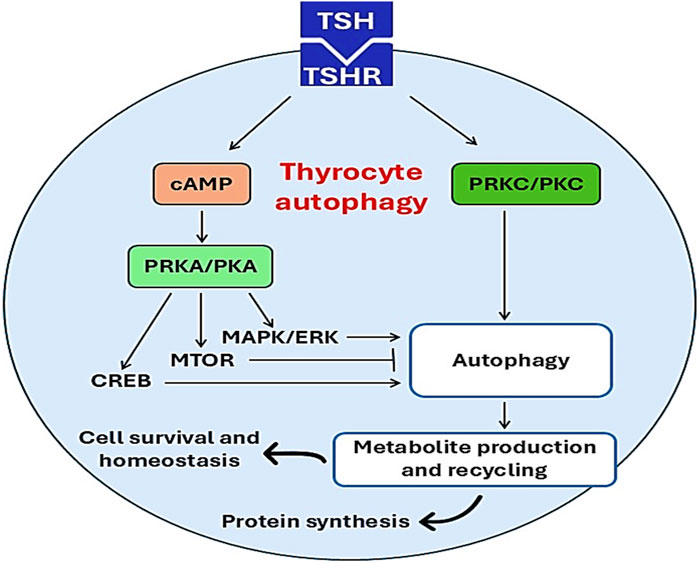
Figure 4. Effects of TSH on thyrocyte autophagy. Activation of TSHR by TSH triggers the activation of thyrocyte autophagy through activation of PKC/cAMP and other signaling pathways. Activated thyrocyte autophagy promotes protein synthesis, cell survival, and nomeostasis.
Conversely, GD-induced hyperthyroidism is associated with augmentation of the circulating levels of T3 and T4 which may affect thyrocyte autophagy. In vivo and ex-vivo findings demonstrate that thyroid hormones stimulate liver fatty acid β-oxidation through induction of autophagy, and blockade of hepatic autophagy by siRNA which targets ATG5 inhibits fatty acid β-oxidation (Sinha et al., 2012). Chi et al. (2019) highlighted that thyroid hormones activate hepatic autophagy in different liver diseases such as non-alcoholic fatty liver disease. Therefore, thyroid hormone-induced autophagy can be a compensatory mechanism to control dysregulated autophagy in GD. Remarkably, polymorphism of the autophagy-related gene IRGM (immunity related GTPase M) is associated with the risk of GD and other AITDs (Yao et al., 2018) signifying a potential link between GD and autophagy dysfunction. IRGM plays a critical role in regulating inflammation by increasing engulfment of apoptotic cells by autophagy (Yao et al., 2018). A case-control study showed that the T allele of rs10065172, A allele of rs4958847, and C allele of rs13361189 are higher in GD patients (Yao et al., 2018). Furthermore, exaggeration of thyroid hormones in GD is linked with the development of oxidative stress, which activates the autophagy flux (Kiffin et al., 2004; Londzin-Olesik et al., 2020). Of interest, oxidative stress is strongly implicated in the pathogenesis of GD (Žarković, 2012). For example, oxidative stress markers are higher in GD patients compared to controls (Ademoğlu et al., 2006). Many studies indicate that oxidative stress through activation of NFKB triggers the development of an autoimmune response in hyperthyroidism (Nandakumar et al., 2008; Makay et al., 2009). NFKB is essential for activation of autophagy (Min et al., 2018) and increases GD risk by 39% (Niyazoglu et al., 2014). Supporting this claim, treatment with the antioxidant selenium reduces the disease severity in GD patients (Marcocci et al., 2011).
These findings proposed that thyrocyte autophagy is dysregulated in GD due to direct effects of TSI and thyroid hormones, and indirectly by oxidative stress and NFKB activation. Moreover, thyrocyte autophagy seems to be inhibited in early GD and activated in late GD to mitigate the inflammatory and oxidative stress disorders.
Autophagy and GD ophthalmopathyGD ophthalmopathy (GO) is the most common extrathyroidal manifestation of GD characterized by unilateral (10%) or bilateral (90%) eye proptosis. GO develops due to activation of T cells and TSI directed against retro-orbital tissues, which share antigenic epitopes with thyrocytes (Nabi and Rafiq, 2020). GO as an autoimmune disease leads to inflammation and injury of extraocular muscles and orbital adipose tissues (Bahn, 2010). The occurrence of GO may precede GD in 23% of cases, coexist with GD in 39% and follow GD in 37% (Claytor and Li, 2021). T cell-mediated activation and the Th1 immune response are activated in the early stage of GO, although the Th2 immune response and antibody production are stimulated in the late stage (Chen et al., 2023). These immune responses activate orbital inflammation and differentiation of adipocytes and myofibroblasts (Li et al., 2021a). These immunoinflammatory changes trigger autophagy, which may induce beneficial or detrimental effects according to the disease stage.
The potential role of autophagy in GO had been discussed in different studies; however, the precise role of autophagy in GO was not fully elucidated (Yoon et al., 2015a). In early GO there is marked inflammatory reactions, which induce aberrant autophagy activation (Guo et al., 2020). GO-associated inflammation is linked with autophagy activation as evidenced by increases of ATG5 and BECN1 and higher conversion of LC3-I to LC3-II (Yoon et al., 2015a; Guo et al., 2020). It has been illustrated that autophagy promotes adipogenesis in patients with GO. A case-control study confirmed that ATG5, LC3 and SQSTM1 are increased in orbital fat from GO patients compared to controls (Yoon et al., 2015a) proposing that activated autophagy is implicated in the pathogenesis of GO. Therefore, inhibition of autophagy may attenuate the progression of GO. In fact, it has been established that autophagy inhibitors chloroquine or hydroxycholoroquine attenuate adipogenesis by inhibiting autophagy of orbital fibroblasts (Guo et al., 2020). Similarly, the autophagy inhibitor bafilomycin A1 or deletion of ATG5 inhibits adipogenesis in orbital fibroblasts (Yoon et al., 2015b). Moreover, astragaloside and icariin suppress orbital fibroblasts and adipogenesis through inhibition of autophagy (Li et al., 2017; Li et al., 2018). These findings indicate that autophagy inhibitors are helpful in the management of GO.
Conversely, the MTOR inhibitor rapamycin, which activates autophagy, produces beneficial effects against GO (Roos and Murthy, 2019; Zhang et al., 2023). Rapamycin improves ocular restriction by inhibiting the differentiation of ocular myofibroblasts (Roos and Murthy, 2019). Indeed, the MTOR pathway is upregulated in patients with GO resulting in the induction of inflammation, fibrosis, and adipogenesis. Of interest, low-dose rapamycin mitigates diplopia/double vision in patients with refractory GO by inhibiting CD4-induced inflammation in GO (Zhang et al., 2023). Of note, rapamycin is also effective in different autoimmune disorders such as systemic sclerosis, systemic lupus erythematosus and rheumatoid arthritis (Bruyn et al., 2008; Su et al., 2009). Recently, it has been shown that rapamycin is more effective than steroids in the management of GO (Lanzolla et al., 2022). Therefore, autophagy activators may be effective in the management of GO. These findings highlight the fact that autophagy plays a double-edged sword role in the pathogenesis of GO—it may be protective or harmful according to the different stages of GO.
Taken together, autophagy may be protective against thyroid GD, but it has dual protective and harmful effects. Therefore, additional preclinical and clinical studies are recommended in this regard.
ConclusionGD is the most common autoimmune disease of the thyroid gland and is characterized by hyperthyroidism and goiter due to production of TSI. TSI activates TSH receptors of the thyroid gland resulting in excessive release of thyroid hormones with subsequent development of hyperthyroidism and goiter. Of note, autophagy plays a critical role in many thyroid diseases and in different stages of the same disease through modulation of immunity and the inflammatory response. In addition, autophagy is also implicated in the pathogenesis of TAO. Thus this review has focused on how autophagy is involved in the pathogenesis of GD regarding its protective and harmful effects. Thyrocyte autophagy is dysregulated in GD due to direct effects of TSI and thyroid hormones, and indirectly by oxidative stress and NFKB activation. Moreover, thyrocyte autophagy seems to be inhibited in early GD and activated in late GD to mitigate the inflammatory and oxidative stress disorders. Importantly, autophagy plays a double-edged sword role in the pathogenesis of GO, where it may be protective or harmful according to the different stages of the disease. Further preclinical and clinical studies are recommended in this regard.
Author contributionsHA-K: Conceptualization, Writing–original draft, Writing–review and editing. GS: Conceptualization, Writing–original draft, Writing–review and editing. HM: Software, Writing–review and editing. MA-A: Investigation, Writing–review and editing. SA: Data curation, Writing–review and editing. MJ: Investigation, Writing–review and editing. AA: Writing–original draft, Writing–review and editing. AA-G: Writing–original draft, Writing–review and editing. DK: Conceptualization, Writing–original draft, Writing–review and editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors are thankful to the Deanship of Graduate Studies and Scientific Research at University of Bisha for supporting this work through the Fast-Track Research Support Program.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AbbreviationsAITD, autoimmune thyroid disease; GD, Graves disease; GO, GD ophthalmopathy; TAO, thyroid-associated opthalmopathy; Th1, type 1 T helper; TSH, thyroid stimulating hormone; TSIs, thyroid-stimulating immunoglobulins.
ReferencesAdemoğlu, E., Özbey, N., Erbil, Y., Tanrikulu, S., Barbaros, U., Yanik, B. T., et al. (2006). Determination of oxidative stress in thyroid tissue and plasma of patients with Graves’ disease. Eur. J. Intern. Med. 17, 545–550. doi:10.1016/j.ejim.2006.04.013
PubMed Abstract | CrossRef Full Text | Google Scholar
AlAnazi, F. H., Al-Kuraishy, H. M., Alexiou, A., Papadakis, M., Ashour, M. H. M., Alnaaim, S. A., et al. (2023). Primary hypothyroidism and alzheimer’s disease: a tale of two. Cell. Mol. Neurobiol. 43, 3405–3416. doi:10.1007/s10571-023-01392-y
PubMed Abstract | CrossRef Full Text | Google Scholar
Ali, N. H., Al-Kuraishy, H. M., Al-Gareeb, A. I., Alnaaim, S. A., Alexiou, A., Papadakis, M., et al. (2023). Autophagy and autophagy signaling in Epilepsy: possible role of autophagy activator. Mol. Med. 29, 142. doi:10.1186/s10020-023-00742-2
PubMed Abstract | CrossRef Full Text | Google Scholar
Al-Kuraishy, H. M., Al-Bdulhadi, M. H., and Al-Gareeb, A. I. (2021). Neuropeptide Y-Agouti related peptide ratio (NAR) in patients with idiopathic primary hypothyroidism: nudge and Risk. J. Pak Med. Assoc. 71 (Suppl. 8), S27–S31.
Al-Kuraishy, H. M., Jabir, M. S., Al-Gareeb, A. I., Klionsky, D. J., and Albuhadily, A. K. (2024a). Dysregulation of pancreatic β-cell autophagy and the risk of type 2 diabetes. Autophagy 18, 2361–2372. doi:10.1080/15548627.2024.2367356
PubMed Abstract | CrossRef Full Text | Google Scholar
Al-Kuraishy, H. M., Jabir, M. S., Al-Gareeb, A. I., Saad, H. M., Batiha, G. E.-S., and Klionsky, D. J. (2024b). The beneficial role of autophagy in multiple sclerosis: yes or no? Autophagy 20, 259–274. doi:10.1080/15548627.2023.2259281
PubMed Abstract | CrossRef Full Text | Google Scholar
Al-Kuraishy, H. M., Sulaiman, G. M., Jabir, M. S., Mohammed, H. A., Al-Gareeb, A. I., Albukhaty, S., et al. (2024c). Defective autophagy and autophagy activators in myasthenia gravis: a rare entity and unusual scenario. Autophagy 20, 1473–1482. doi:10.1080/15548627.2024.2315893
PubMed Abstract | CrossRef Full Text | Google Scholar
Antonelli, A., Fallahi, P., Elia, G., Ragusa, F., Paparo, S. R., Ruffilli, I., et al. (2020). Graves’ disease: clinical manifestations, immune pathogenesis (cytokines and chemokines) and therapy. Best Pract. and Res. Clin. Endocrinol. and Metabolism 34, 101388. doi:10.1016/j.beem.2020.101388
PubMed Abstract | CrossRef Full Text | Google Scholar
Brix, T. H., Kyvik, K. O., Christensen, K., and Hegedüs, L. (2001). Evidence for a major role of heredity in Graves’ disease: a population-based study of two Danish twin cohorts. J. Clin. Endocrinol. and Metabolism 86, 930–934. doi:10.1210/jcem.86.2.7242
PubMed Abstract | CrossRef Full Text | Google Scholar
Broggi, G., Ieni, A., Russo, D., Varricchio, S., Puzzo, L., Russo, A., et al. (2020). The macro-autophagy-related protein Beclin-1 immunohistochemical expression correlates with tumor cell type and clinical behavior of uveal Melanoma. Front. Oncol. 10, 589849. doi:10.3389/fonc.2020.589849
PubMed Abstract | CrossRef Full Text | Google Scholar
Bruyn, G. A. W., Tate, G., Caeiro, F., Maldonado-Cocco, J., Westhovens, R., Tannenbaum, H., et al. (2008). Everolimus in patients with rheumatoid arthritis receiving concomitant methotrexate: a 3-month, double-blind, randomised, placebo-controlled, parallel-group, proof-of-concept study. Ann. rheumatic Dis. 67, 1090–1095. doi:10.1136/ard.2007.078808
PubMed Abstract | CrossRef Full Text | Google Scholar
Cao, J., Su, Y., Chen, Z., Ma, C., and Xiong, W. (2022). The risk factors for Graves’ ophthalmopathy. Graefe’s Archive Clin. Exp. Ophthalmol. 260, 1043–1054. doi:10.1007/s00417-021-05456-x
PubMed Abstract | CrossRef Full Text | Google Scholar
Cao, W., Li, J., Yang, K., and Cao, D. (2021). An overview of autophagy: mechanism, regulation and research progress. Bull. Du. cancer 108, 304–322. doi:10.1016/j.bulcan.2020.11.004
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, Y.-Q., Gao, L.-D., Liu, Y.-L., Shen, Y., Diao, J.-L., Yang, W.-H., et al. (2023). Autophagy in graves’ ophthalmopathy. Front. Cell. Dev. Biol. 11, 1158279. doi:10.3389/fcell.2023.1158279
PubMed Abstract | CrossRef Full Text | Google Scholar
Chi, H.-C., Tsai, C.-Y., Tsai, M.-M., Yeh, C.-T., and Lin, K.-H. (2019). Molecular functions and clinical impact of thyroid hormone-triggered autophagy in liver-related diseases. J. Biomed. Sci. 26, 24–15. doi:10.1186/s12929-019-0517-x
PubMed Abstract | CrossRef Full Text | Google Scholar
Davies, T. F., Andersen, S., Latif, R., Nagayama, Y., Barbesino, G., Brito, M., et al. (2020). Graves’ disease. Nat. Rev. Dis. Prim. 6, 52. doi:10.1038/s41572-020-0184-y
PubMed Abstract | CrossRef Full Text | Google Scholar
Duan, J., Deng, T., Kang, J., and Chen, M. (2019). DINP aggravates autoimmune thyroid disease through activation of the Akt/mTOR pathway and suppression of autophagy in Wistar rats. Environ. Pollut. 245, 316–324. doi:10.1016/j.envpol.2018.10.108
PubMed Abstract | CrossRef Full Text | Google Scholar
Fang, S., Lu, Y., Huang, Y., Zhou, H., and Fan, X. (2021). Mechanisms that underly T cell immunity in graves’ orbitopathy. Front. Endocrinol. 12, 648732. doi:10.3389/fendo.2021.648732
PubMed Abstract | CrossRef Full Text | Google Scholar
Faustino, L. C., Lombardi, A., Madrigal-Matute, J., Owen, R. P., Libutti, S. K., and Tomer, Y. (2018). Interferon-α triggers autoimmune thyroid diseases via lysosomal-dependent degradation of thyroglobulin. J. Clin. Endocrinol. and Metabolism 103, 3678–3687. doi:10.1210/jc.2018-00541
PubMed Abstract | CrossRef Full Text | Google Scholar
Ferrari, S. M., Fallahi, P., Ruffilli, I., Elia, G., Ragusa, F., Benvenga, S., et al. (2019). The association of other autoimmune diseases in patients with Graves’ disease (with or without ophthalmopathy): review of the literature and report of a large series. Autoimmun. Rev. 18, 287–292. doi:10.1016/j.autrev.2018.10.001
PubMed Abstract | CrossRef Full Text | Google Scholar
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo-San Pedro, J. M., Cecconi, F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. doi:10.15252/embj.201796697
PubMed Abstract | CrossRef Full Text | Google Scholar
Guo, Y., Li, H., Chen, X., Yang, H., Guan, H., He, X., et al. (2020). Novel roles of chloroquine and hydroxychloroquine in Graves’ orbitopathy therapy by targeting orbital fibroblasts. J. Clin. Endocrinol. and Metabolism 105, 1906–1917. doi:10.1210/clinem/dgaa161
PubMed Abstract | CrossRef Full Text | Google Scholar
Hargreaves, C. E., Grasso, M., Hampe, C. S., Stenkova, A., Atkinson, S., Joshua, G. W. P., et al. (2013). Yersinia enterocolitica provides the link between thyroid-stimulating antibodies and their germline counterparts in Graves’ disease. J. Immunol. 190, 5373–5381. doi:10.4049/jimmunol.1203412
PubMed Abstract | CrossRef Full Text | Google Scholar
He, K., Jiang, P., Liu, B., Liu, X., Mao, X., and Hu, Y. (2020). Intrathyroid injection of dexamethasone inhibits Th2 cells in Graves’ disease. Archives Endocrinol. metabolism 64, 243–250. doi:10.20945/2359-3997000000244
PubMed Abstract | CrossRef Full Text | Google Scholar
Hussain, Y. S., Hookham, J. C., Allahabadia, A., and Balasubramanian, S. P. (2017). Epidemiology, management and outcomes of Graves’ disease—real life data. Endocrine 56, 568–578. doi:10.1007/s12020-017-1306-5
PubMed Abstract | CrossRef Full Text | Google Scholar
Inaba, H., De Groot, L. J., and Akamizu, T. (2016). Thyrotropin receptor epitope and human leukocyte antigen in Graves’ disease. Front. Endocrinol. 7, 218181. doi:10.3389/fendo.2016.00120
PubMed Abstract | CrossRef Full Text | Google Scholar
Iriondo, M. N., Etxaniz, A., Varela, Y. R., Ballesteros, U., Lázaro, M., Valle, M., et al. (2023). Effect of ATG12–ATG5-ATG16L1 autophagy E3-like complex on the ability of LC3/GABARAP proteins to induce vesicle tethering and fusion. Cell. Mol. Life Sci. 80, 56. doi:10.1007/s00018-023-04704-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Kiffin, R., Christian, C., Knecht, E., and Cuervo, A. M. (2004). Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell. 15, 4829–4840. doi:10.1091/mbc.e04-06-0477
PubMed Abstract | CrossRef Full Text | Google Scholar
Kurashige, T., Nakajima, Y., Shimamura, M., Matsuyama, M., Yamada, M., Nakashima, M., et al. (2019). Basal autophagy deficiency causes thyroid follicular epithelial cell death in mice. Endocrinology 160, 2085–2092. doi:10.1210/en.2019-00312
PubMed Abstract | CrossRef Full Text | Google Scholar
Kurashige, T., Nakajima, Y., Shimamura, M., Yamada, M., and Nagayama, Y. (2020). Hormonal regulation of autophagy in thyroid PCCL3 cells and the thyroids of male mice. J. Endocr. Soc. 4, bvaa054. doi:10.1210/jendso/bvaa054
PubMed Abstract | CrossRef Full Text | Google Scholar
Lanzolla, G., Maglionico, M. N., Comi, S., Menconi, F., Piaggi, P., Posarelli, C., et al. (2022). Sirolimus as a second-line treatment for Graves’ orbitopathy. J. Endocrinol. Investigation 45, 2171–2180. doi:10.1007/s40618-022-01862-y
PubMed Abstract | CrossRef Full Text | Google Scholar
Leporati, P., Groppelli, G., Zerbini, F., Rotondi, M., and Chiovato, L. (2015). Etiopathogenesis of Basedow's disease. Trends and current aspects. Nuklearmedizin-NuclearMedicine 54, 204–210. doi:10.3413/Nukmed-0739-15-04
PubMed Abstract | CrossRef Full Text | Google Scholar
Lesmana, R., Sinha, R. A., Singh, B. K., Zhou, J., Ohba, K., Wu, Y., et al. (2016). Thyroid hormone stimulation of autophagy is essential for mitochondrial biogenesis and activity in skeletal muscle. Endocrinology 157, 23–38. doi:10.1210/en.2015-1632
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, H., Gao, L., Min, J., Yang, Y., and Zhang, R. (2021a). Neferine suppresses autophagy-induced inflammation, oxidative stress and adipocyte differentiation in Graves’ orbitopathy. J. Cell. Mol. Med. 25, 1949–1957. doi:10.1111/jcmm.15931
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, H., Yuan, Y., Zhang, Y., Zhang, X., Gao, L., and Xu, R. (2017). Icariin inhibits AMPK-dependent autophagy and adipogenesis in adipocytes in vitro and in a model of graves’ orbitopathy in vivo. Front. physiology 8, 223122. doi:10.3389/fphys.2017.00045
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, H., Zhang, Y., Min, J., Gao, L., Zhang, R., and Yang, Y. (2018). Astragaloside IV attenuates orbital inflammation in Graves’ orbitopathy through suppression of autophagy. Inflamm. Res. 67, 117–127. doi:10.1007/s00011-017-1100-0
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, Q. M., Wei, J. P., Li, M., and Meng, S. H. (2015). Effect of jiakangning capsule on thyroid function and akt/mTOR signal pathway of graves’ disease mice: an experimental study. Chin. J. Integr. Traditional West. Med. 35, 1119–1124.
PubMed Abstract | Google Scholar
Li, W., He, P., Huang, Y., Li, Y.-F., Lu, J., Li, M., et al. (2021b). Selective autophagy of intracellular organelles: recent research advances. Theranostics 11, 222–256. doi:10.7150/thno.49860
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, Y., Zhang, W., Liu, M., Zhang, Q., Lin, Z., Jia, M., et al. (2021c). Imbalance of autophagy and apoptosis induced by oxidative stress may be involved in thyroid damage caused by sleep deprivation in rats. Oxidative Med. Cell. Longev. 2021, 2021. doi:10.1155/2021/5645090
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, Y., Zhao, C., Zhao, K., Yu, N., Li, Y., Yu, Y., et al. (2021d). Glycosylation of anti-thyroglobulin IgG1 and IgG4 subclasses in thyroid diseases. Eur. Thyroid J. 10, 114–124. doi:10.1159/000507699
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, Y., Yuan, X., Li, X., Cui, D., and Xie, J. (2018). Constitutive changes in circulating follicular helper T cells and their subsets in patients with Graves’ disease. J. Immunol. Res. 2018, 8972572. doi:10.1155/2018/8972572
PubMed Abstract | CrossRef Full Text | Google Scholar
Londzin-Olesik, M., Kos-Kudła, B., Nowak, A., Wielkoszyński, T., and Nowak, M. (2020). The effect of thyroid hormone status on selected antioxidant parameters in patients with Graves’ disease and active thyroid-associated orbitopathy. Endokrynol. Pol. 71, 418–424. doi:10.5603/EP.a2020.0049
留言 (0)