
記住我


The trial is designed as an observer-blind randomized controlled study, which was conducted from October 2023 to March 2024. This study was approved by medical ethics committee, and all patients provided informed consent. This study was approved by the Medical Ethics Committee of Weifang People’s Hospital (KYLL20230908-4), and the trial was registered with the Chinese Clinical Trial Register (Registration number. ChiCTR2300076237). This study was conducted in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
In the design, analysis, and reporting of this trial, we adhered strictly to the Consolidated Standards of Reporting Trials (CONSORT) guidelines [24].
PatientsIn our center, when patients were diagnosed with lumbar spinal stenosis, patients who were willing to undergo surgical treatment were included in the experimental cohort. Therefore, patients who planned to undergo minimally invasive surgical treatment for lumbar spinal stenosis were prospectively screened by our medical team from September 2023 to December 2023for inclusion in this trial. The inclusion criteria were as follows: (1) Radiographic confirmation of lumbar spinal stenosis; (2) Presence of characteristic neurogenic intermittent claudication with bilateral lower limb neurologic deficits, and lack of significant improvement after 3 months of conservative management; (3) Age are ranged from 18 to 80 years; (4) Ability to provide informed consent, with capacity to read and comprehend the informed consent form independently or with family assistance, as well as understand and adhere to follow-up questionnaires; (5) Willingness to consent to and fully comply with the clinical protocol, including adherence to the follow-up schedule and requirements. The exclusion criteria included (1) Lumbar spinal stenosis due to fracture, tumor, or infection; (2) History of spinal surgery; (3) Complete limb paralysis; (4) Contraindications to surgery or refusal of surgical intervention; (5) Presence of psychiatric or psychological disorders, such as schizophrenia, depression, history of chronic opioid use, alcohol abuse, or opioid dependence; (6) Participation in other clinical trials ; (7) Loss to follow-up; (8) Due to various reasons for unblinding; (9) Other conditions deemed unsuitable for participation in this study by the investigators.
Double-blind prospective randomized controlled trialOutpatient imaging and physical assessments were employed to determine patient eligibility. Patients who satisfied the inclusion and exclusion criteria based on these assessments were provided with a comprehensive explanation of the trial’s process and objectives to ensure fully informed consent. However, they were blinded to the specific study intervention (Delta large-channel endoscopy or UBE). A statistician generated random numbers from 001 to 101 specifically for this trial using SPSS software version 26.0, with each patient assigned a unique random number. These numbers were placed in sealed envelopes and securely stored by our center staff. Upon confirming a patient’s eligibility, the sealed envelope was opened, and the surgeon was notified of the patient’s assignment. Eligible patients who provided informed consent were then randomized into either the Delta large-channel endoscopy group or the UBE group in a 1:1 ratio according to the predetermined randomization sequence, with odd numbers assigned to the UBE group and even numbers to the Delta large-channel endoscopy group, ensuring complete randomness in the allocation process. During the procedure, open conversion or conversion of one group to another is allowed to ensure the rights and interests of the patient, but the relevant information must be recorded in detail. Perioperative outcomes were assessed by a single investigator who was blinded to the study intervention until the end of the trial.
Anesthesia and surgical proceduresAll patients included in Delta large-channel endoscopy group or UBE group underwent total intravenous anesthesia. Identical monitoring, anesthesia, and analgesic regimens were used in both the Delta large-channel endoscopy group and the UBE group. Continuous blood pressure, heart rate, peripheral pulse oximetry, bispectral index (BIS, Covidien/Medtronic), and electrocardiography monitoring were performed using standard monitoring equipment for both groups. Before the procedures, intravenous (IV) midazolam (0.03 mg/kg), IV sufentanil (0.3–0.4 µg/kg), IV propofol (1.5–2 mg/kg), and IV cisatracurium (0.2 mg/kg) were administered for induction. Mechanical ventilation was facilitated by endotracheal intubation. Continuous intravenous infusion of propofol and remifentanil was used for maintenance. Vasoactive drugs were administered as needed to maintain heart rate and mean arterial pressure within 20% of baseline. Supplementary intraoperative analgesics were not administered. Close monitoring and recording of all intraoperative physical parameters, including fluid input and output and drug dosages, were performed. After the procedure, patients are prescribed Paracetamol and Dihydrocodeine Tartrate Tablets (acetaminophen 500 mg: dihydrocodeine tartrate 10 mg), to be taken one tablet every six hours for three days. During the perioperative period, all patients did not use other pain management methods such as patient-controlled analgesia or nerve block.
Delta large-channel endoscopy groupThe patient was placed prone under tracheal intubation general anesthesia. Using the C-arm, the relevant vertebrae were located and the skin surface marked. The lamina and facet joint were separated, and the Delta large-channel endoscopy working channel inserted. Fluoroscopy confirmed the operation level. The posterior lens system was set up, and saline irrigation continued. The lower edge of the upper lamina and the lower articular process were exposed; bone was removed using a ring saw or grinding drill. Hemostasis was achieved with a plasma radiofrequency knife. The ligamentum flavum was separated, the dura mater and nerve root released, and adhesions removed. The annulus fibrosus was cut, nucleus pulposus tissue extracted, and cartilage on the endplates scraped under direct endoscopic view. Endplate condition was assessed before removing the endoscope; drainage was not routinely performed. Patients are encouraged to begin light activities the next day. After the procedure, patients are instructed to wear waist support from the day after surgery up to one month.
UBE groupThe patient was placed prone under tracheal intubation general anesthesia. Using a C-arm, the target intervertebral space was identified, and a mark was placed 2 cm lateral and 1.0–1.5 cm below this space. A horizontal incision was made at this mark, and through layered dissection, muscles and fascia were cut, exposing the lamina surface. The endoscopic system was then set up, and the surgical area was rinsed clean. A plasma radiofrequency knife was used to achieve hemostasis and prepare the lamina surface, revealing the lamina and facet joint. The lower edge of the upper vertebra’s lamina, the medial side of the articular process, and the upper edge of the lower vertebra’s lamina were excised to access the spinal canal. Adhesions around the nerve were dissected, and the ligamentum flavum was removed. Following another round of hemostasis, the nerve root and dural sac were retracted; the annulus fibrosus was incised, nucleus pulposus removed, and cartilage scraped from the endplates under direct endoscopic view. The endplates were evaluated, the endoscope removed, and a drainage tube was inserted into the decompression area. Patients are encouraged to begin light activities the next day. After the procedure, patients are instructed to wear waist support from the day after surgery up to one month.
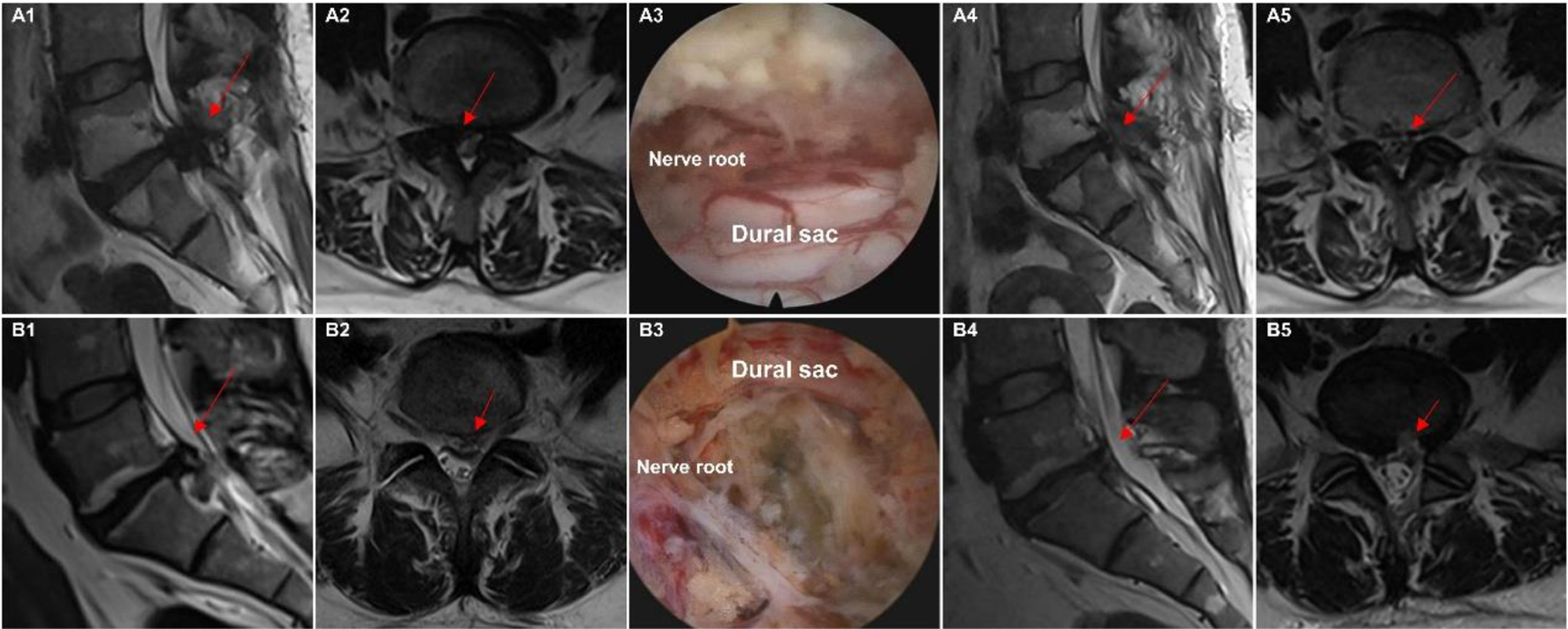
Figure 1 illustrates intraoperative images of the two interventions.
Fig. 1
Intraoperative and pre/postoperative images of the two interventions. Panel (A) shows the Delta large-channel endoscopy group (A1–A5), with preoperative MRI images (A1, A2) depicting lumbar spinal stenosis, followed by an intraoperative image (A3) showing complete decompression of the dural sac. Postoperative MRI images (A4, A5) demonstrate the decompressed spinal canal with improved dural sac area and spinal canal diameter. Panel (B) shows the Unilateral biportal endoscopy group (B1–B5), following the same sequence as described for panel (A)
Primary indicatorsThe primary observation indicators included oswestry disability index (ODI) scores and operation time. The ODI is a specific questionnaire for waist related disability and quality of life (QOL) [25]. Patients completed questionnaires including the ODI before and 14 days, 1, 2, and 3 months after surgery. The ODI is a widely used and validated score of lumbar function. The questionnaire contains 10 questions, each with 6 answers (on a scale from 0 to 5); The ODI total score is the sum of these scores, converted to a 0-100 score, with higher scores indicating greater disability and worse quality of life. The operation time of the surgical procedure was defined as the time from the initial skin incision to the final skin closure.
Secondary indicatorsThe secondary measures included visual analogue scale (VAS), European Quality of Life 5 dimension (EQ-5D) score, Japanese Orthopaedic Association (JOA) score, intraoperative blood loss, length of hospital stay, and hospitalization cost. The VAS score for the low back and leg pain questionnaire contains a 10-cm line with “none” at one end (0) and “severe pain” at the other end (10). VAS questionnaires containing leg pain and low back pain were collected before surgery, on postoperative night, 3 days, 7 days, 14 days, 1 month, 2 months and 3 months after surgery. The highest postoperative JOA total score was 29 and the lowest was 0, with lower scores indicating more significant dysfunction. The EQ-5D score combines responses to a five-dimensional questionnaire, each consisting of three levels, ranging from a score between − 0.59 and 1, with 1.00 indicating full health. EQ-5D scale scores were referenced to their specific set of utility values in the Chinese population [26]. The Japanese Orthopaedic Association (JOA) score is a clinical tool used to evaluate the functional status of patients with spinal disorders [27]. It consists of four parts: pain intensity, motor function, sensory function, and activities of daily living. Scores are calculated by summing the individual component scores, with higher scores indicating better function and lower scores indicating more severe symptoms. Patients completed questionnaires including EQ-5D and JOA before surgery and 14 days, 1, 2, and 3 months after surgery. Length of hospital stay, and hospitalization costs were collected using a Case Record Form (CRF). In addition, the postoperative complications of the two groups were recorded in detail. One of the most frequent complications in spinal surgery, including minimally invasive techniques, is dural tear, which is a rupture of the outer membrane enveloping the spinal cord. Such tears can lead to cerebrospinal fluid (CSF) leakage, causing headaches, infection, and delayed wound healing. Additionally, there is a risk of nerve root injury during minimally invasive spinal surgery that may result in leg weakness, numbness, or pain. Although minimally invasive spinal surgery generally leads to reduced blood loss compared to open surgery, there is still a potential for bleeding and hematoma formation. Postoperative hematomas may exert pressure on the spinal nerves, necessitating surgical intervention in severe cases. Some patients may continue to experience persistent pain or incomplete symptom relief after minimally invasive spinal surgery, requiring revision surgery within three months.
Sample size calculation and statistical analysisIn our previous retrospective study, the standard deviations of the change in ODI scores after the two surgeries were 18.26 and 21.57, respectively. We chose a one-sided alpha level of 0.05, a non-inferiority margin of 0.2, allowed for a 10% loss to follow-up, and the minimum detectable change (MDC) for the ODI score is 12.8 points [28]. From this calculation, if we want to have 80% power to demonstrate the non-inferiority of Delta large-channel endoscopy compared to UBE, each group needs at least 41 patients [29]. We therefore planned to enroll 50 patients per group to increase confidence in the results.
The Shapiro-Wilk test was employed to evaluate the distribution type of the data. Continuous data with normal distribution in both groups were presented as mean ± standard deviation (SD) and compared using an unpaired independent sample t-test. Continuous data not following a normal distribution were expressed as median (range) or median (25th and 75th percentiles) and compared using the Mann-Whitney U test. Categorical data were summarized as frequencies and percentages and compared using Pearson’s chi-square test or Fisher’s exact test. For primary outcomes, the operation time was analyzed using an independent sample t-test, while ODI scores at each postoperative follow-up time point (14 days, 1 month, 2 months, and 3 months) were also analyzed using this method. Intergroup differences in ODI scores across the four postoperative time points were further examined using repeated measures ANOVA. Secondary clinical outcomes (VAS pain scores for the back and lower extremities, JOA scores, and EQ-5D scores) were analyzed using an independent sample t-test at each postoperative follow-up time point, and intergroup differences across these time points were examined using repeated-measures ANOVA. Time was treated as a categorical variable with an intervention-by-time interaction to assess the intervention effect at each follow-up time point. Between-group differences over 3 months were analyzed, controlling for the follow-up time point as a categorical variable. A p-value < 0.05 was deemed statistically significant. Statistical analyses were conducted using SPSS 26.0 (IBM Statistics, Armonk, NY) and GraphPad Prism 10.0 (GraphPad Software, San Diego, CA, USA).
留言 (0)