
記住我


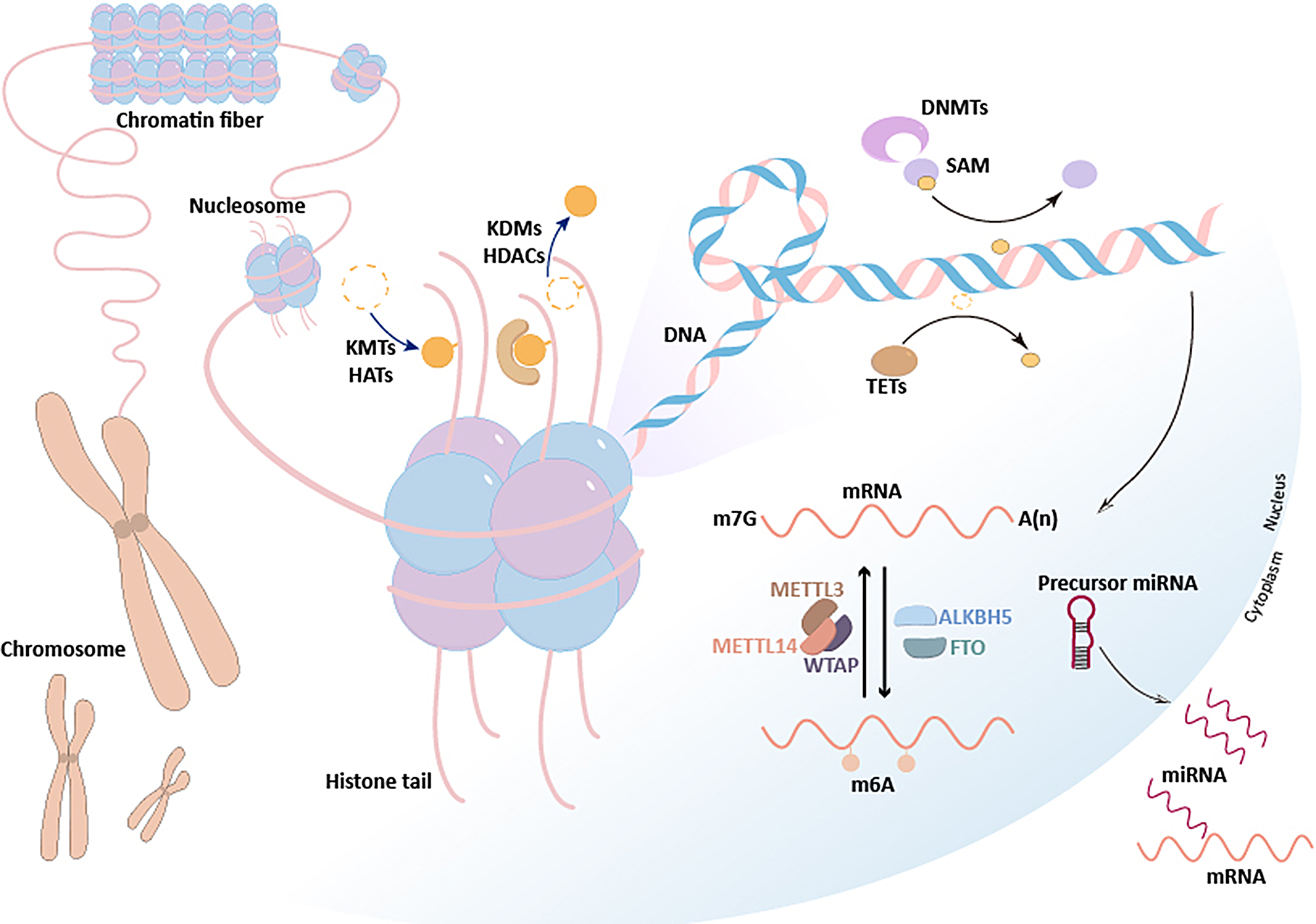






One of the many important processes involved in epigenetics is the modification of DNA structure through adding chemical residues, such as methyl, carboxyl, hydroxymethyl, dimethyl and many others, to one of the four standard nucleotides: adenine (A), cytosine (C), guanine (G), thymine (T). The most studied type of such modifications is DNA methylation, which appears when a methyl group is added to C or A within the DNA [28].
During early embryogenesis, there is an epigenetic reprogramming consisting of the development of global DNA demethylation and remethylation [29], which indicates limited evidence for transgenerational inheritance of DNA methylation in mammalian systems [8, 30]. Nevertheless, it has been hypothesized that environmental exposures are able to modify the epigenetic control on gene expression, and that the phenotype is the result of the interaction between genotype and environment. This explains why the methylome has proved to be dynamic during development, cell differentiation and aging [30,31,32]. Identifying DNA methylation in the genome is very important for understanding the purposes of epigenetic modifications [6].
Almost half of the human genome (~ 45%) consists of silenced viral and transposable elements, which display high levels of DNA methylation. Therefore, methylation is essential, as the expression of these harmful elements could lead to gene disruption and mutations [12]. Nevertheless, some of these TEs have been co-opted by the host to perform essential regulatory functions (e.g. TEs are upregulated during early development and in the neuronal lineage and dysregulations have been proved to be involved in the development of neurological disorders and cancer [33]).
The base pairing of cytosine and guanine occurs through phosphate (p) links, therefore the abbreviated form of C-G dinucleotides is CpG (cytosine-phosphate-guanine). CpGs do not have an even distribution throughout the genome, as they tend to assemble in their unmethylated form in areas called CpG islands (CGIs), which are usually associated with gene promoters [13]. The rest of the CpG sites found throughout the human genome (70–80%) are methylated, predominantly as 5mC [18], and they transform into thymine through deamination over time [34].
Apart from 5mC and 5hmC, there are other forms of methylation present throughout the human genome, such as N6-methyladenine (6 mA, involved in the development of different types of neoplasia, such as gastric and non-small-cell lung cancer [35, 36]), N4-methylcytosine (4mC, involved in the development of gastrointestinal cancer [37]), and the oxidation products 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC, possibly involved in the development of prostate cancer) [6, 18, 38].
The chemistry of DNA methylation: DNA methyltransferase enzymesDNA methylation is possible due to DNA methyltransferase enzymes (DNMTs) which are able to transfer a methyl group from S-adenosyl methionine (SAM) to the fifth carbon of C within the DNA to form 5mC [12], fulfilling an essential role in embryogenesis [13, 39]. The first DNMT (DNMT1) was discovered in 1988 [40], followed by DNMT3a and DNMT3b in 1998 [41]. Recently, DNMT3L has been described as a cofactor, while in 2016, DNMT3C has been discovered as a new methyltransferase involved mostly in fertility by protecting male germinative cells from the activity of transposons [42]. These are not the only important enzymes involved in the methylation process. They can actually be categorized in 3 groups of enzymes: the writers, erasers, and readers. DNMTs are writers, catalyzing the addition of methyl to C residues. Erasers, such as the ten-eleven translocation (TET) enzymes [12] which have been discovered in 2010 [43], and the activation-induced cytidine deaminase (AID) / apolipoprotein B messenger ribonucleic acid (mRNA) editing enzyme, catalytic polypeptide (APOBEC), are responsible for demethylation, which consists of removing the methyl groups from DNA. Readers, such as methyl-CpG-binding domain (MBD) proteins, ubiquitin-like, containing PHD and RING finger domains (UHRF) proteins, and zinc-finger proteins, are able to bind methyl groups in order to influence the expression of genes [12].
DNMTs fulfill complementary roles in order to maintain the methylation patterns in mammals. DNMT1, DNMT3a, DNMT3b and DNMT3c have different, but essential purposes [18, 42] (see Fig. 1).
Fig. 1
The roles of DNA methyltransferase enzymes (DNMTs) [12, 18, 42]
DNMT1 is known as ‘the maintenance enzyme’, which ensures that the methylation pattern is preserved between cell divisions. It is the best studied DNMT, and it is present in high concentrations in dividing cells, as it has an affinity for the hemimethylated DNA present at the replication fork during DNA replication. It has the ability to bind to the newly synthesized DNA strand and it adds methyl groups according to the prior pattern.
DNMT3a and DNMT3b are known as ‘de novo methyltransferases’, because they add methyl groups to unmethylated C within the DNA strands. They have a similar structure, as for the functions, DNMT3a tends to be ubiquitously expressed and it is important for cell differentiation and maternal imprinting, while DNMT3b has a low expression in differentiated tissues, except for bone marrow, testes and the thyroid gland, and it is crucial in early development and X-chromosome inactivation in females [12, 18]. DNMT3L has recently been discovered as a member of the DNMT family lacking a catalytic domain, which is able to stimulate the enzymatic activity of DNMT3a and DNMT3b, while being expressed mostly in early development [12, 44, 45].
Demethylation (mechanism of methyl groups removal) can occur passively through a loss of maintenance during cell division, or actively through eraser enzymes such as TET enzymes (TET1, TET2, TET3), which are able to transform 5mC groups into 5-hydroxymethylcytosine (5hmC) [13, 30]. Numerous 5hmC residues are present in the developed brain, and it is unclear if 5hmC is only an intermediate step in DNA demethylation or if it has its own epigenetic roles in gene expression [12, 46]. The inhibition of methylation enzymes (DNMTs) with different molecules such as azacitidine and decitabine (drugs used in oncology), can be useful in modifying the malignant phenotype of the cells through re-expression of tumor suppressor genes [8, 47, 48].
Roles of DNA methylationThe process of DNA methylation is essential for normal development in mammals, having important roles in early embryonic development, stem cell differentiation, tissue maturation, and whole genome studies have proved that methylation is cell-type specific [30, 49]. However, cell-to-cell epigenetic variations have been identified within homogenous cell populations, therefore DNA methylation could be considered an important factor in biological variability of malignant tumors [6, 50].
Many physiopathological processes, both normal and abnormal, have been associated with DNA methylation (especially 5mC) (see Fig. 2): X chromosome inactivation, genomic imprinting, chromosome stability [18] and structure modulation, transposon activation, inflammation [6], genome integrity and stability, poly(A) tail length regulation [1, 51], silencing repetitive DNA [13]. Effects on RNA splicing, degradation and translation have been associated with the presence of 6mA [1]. Some of the pathological processes linked to methylation are: malignancy, imprinting disorders (Angelman syndrome, Prader-Willi syndrome, Beckwith-Wiedemann syndrome, Silver-Russell syndrome), X-chromosomal recessive disorders (e.g. Duchenne’s muscular dystrophy, Haemophilia B), trinucleotide repeat disorders (e.g. Fragile X syndrome, Friedreich ataxia), defects in gene expression regulation machinery (e.g. Immunodeficiency, Centromere instability and Facial anomalies syndrome (ICF syndrome), Alpha thalassemia X-linked intellectual disability (ATRX syndrome), Rett syndrome) [8], as well as phenotypes of developmental delays and congenital anomalies which have yet been attributed a specific genetic cause [52].
Epimutations can be defined as random errors of the epigenetic machinery, which can be associated with a multitude of environmental factors (e.g. tobacco smoking, foods and dietary factors) that have effects on DNA methylation [8, 31].
Fig. 2
Physiopathological processes in which DNA methylation is involved. The physiological processes are colored in green, while the pathologies are colored in red. Abbreviations: Immunodeficiency, centromere instability and facial anomalies syndrome (ICF syndrome), Alpha thalassemia X-linked intellectual disability (ATRX syndrome)
X-chromosome inactivationDNA methylation is responsible for the inactivation of one of the two X-chromosomes present in females [8], through high rates of methylation found in the proximity of promoter regions. Normally, in this area, the DNA tends to be unmethylated in order to allow the actions of gene promoters; however, when DNA methylation occurs, the genes are silenced and the respective X-chromosome is inactivated [18]. This process is coordinated in early development by the long non-coding RNA (lncRNA) XIST, which is first transcribed and afterwards it spreads in cis across the inactive X-chromosome. It has been thought that this is the only role of XIST, but recently it has been discovered that XIST is still needed in adult human B-cells for silencing X-linked immune genes (e.g. TLR7) through XIST-dependent histone deacetylation, since these genes lack promoter DNA methylation [53].
ImprintingRecently, DNA methylation analysis from blood samples has become the first diagnostic procedure in the management of patients with a suspicion of imprinting disorders [8]. Gene regulation through methylation of CGIs is an important step for imprinting [12]. Usually, most gene transcripts are expressed from both parental alleles: maternal and paternal. Imprinted genes are genes expressed only from the maternal or the paternal allele, based on imprinting control regions: these are parent-of-origin (PofO) differentially methylated regions (DMRs) [7]. There are hundreds of differentially methylated loci in the human genome, according to the PofO, which are quite constant across tissues, individuals and populations. The differential methylation occurs in gametes or after fertilization, and it persists in adults [54], fulfilling essential roles in both imprinting, and embryogenesis [55]. DMRs can also have crucial roles in determining phenotypes (e.g. altering the expression of mismatch repair genes could lead to malignant growth) [49].
Allele-specific methylation patterns are difficult to identify through short-read sequencing. In 2012, Fang F. et al. have developed a probabilistic model, independent of genotype, in order to identify allele-specific methylation patterns based on data obtained from bisulfite sequencing, by describing how the methylation state of each read reflects two distinct patterns which contain half the data. This was a first step in integrating computational strategies in order to enhance the accuracy of detecting allele-specific methylation patterns [56]. In 2015, progress was made in this specific area through a method called Pyrosequencing, a real-time sequencing method which was able to analyze the patterns of methylation separately on each allele. It was able to identify individuals heterozygous for a SNP from a region of interest, and the bisulfite-treated DNA was then analyzed for identifying regions of potential allele-specific DNA methylation surrounding that specific SNP. The newly discovered DNA methylation patterns were then individually amplified using allele-specific PCR in order to be further analyzed in a more detailed matter [57]. Currently, an easier and more efficient method for identifying allele-specific methylation patterns is long-read sequencing, through nanopore technology [7, 49].
Alterations of imprinting are responsible for many diseases: Prader-Willi Syndrome, Angelman Syndrome, Beckwith-Wiedemann Syndrome, and cancer [55], and the diagnosis for known imprinting disorders can be made through simultaneous screening [58]. Prader-Willi syndrome appears through loss of the paternal allele of chromosome 15q11-q13 [8 ]and it is characterized by hypotonia in infants, followed by obesity and excessive eating after early childhood, associated with significant mental impairments [12, 58]. The loss of the maternal allele of chromosome 15q11-q13 causes Angelman syndrome [88], associated with epilepsy, intellectual disability, limited speech and truncal ataxia [58]. Silver-Russell-syndrome occurs through a somatic mosaic defect and it is characterized by severe intrauterine and postnatal growth impairments. The same mechanism applies to Beckwith-Wiedemann syndrome, which is associated with overgrowth, malformations and predisposition to tumors [8, 58].
Repetitive sequences-associated diseasesTransposable elements (TEs) are DNA elements that change chromosomal position and copy number within a genome. There are two main classes of TEs, namely: (i) DNA transposons that use a cut-and-paste mechanism and (ii) retrotransposons that replicate their DNA copies through an RNA intermediate. Integration of TEs either through cut-and-paste or retrotransposition can happen either inside genes (affecting the sequence of the gene) or in the vicinity of genes (which can lead to abnormal activation or repression of the neighboring genes) [59]. Transposable elements can be directly associated with a number of diseases, such as hemophilia (which could be considered as the prototype for a disease-causing insertion), muscular dystrophy, various types of cancer [60] (cancers of the pancreas, colon, ovaries [59], breast [61], bladder [62], liver [63], B-cell non-Hodgkin lymphoma [64]), senescence of mesenchymal progenitor cells [
留言 (0)