
記住我







The cunner reference genome, included in the Vertebrate Genomes Project [54], was screened for AFP sequences using the cunner cDNA sequence [29]. Matches were found at a single location, spanning 133 kbp, on chromosome 4 (Fig. 2A). This genome was not annotated, so the eleven AFP genes found here were annotated based on the known cDNA sequence (Fig. 2A, cyan arrowheads). Additionally, seven interspersed (yellow and red arrowheads) and five flanking genes (grey arrowheads) in the immediate neighborhood were also identified and marked based upon the annotated genomes of the spotty and ballan wrasses [54], two closely related fishes in the same family (Labridae, commonly called wrasses). The microsynteny of the flanking genes is conserved among the three species (Fig. 2, grey arrows), with the five encoded proteins sharing 89–99% identity between the cunner and ballan wrasse. As expected, the identities between the cunner/ballan wrasse and the more distantly related spotty are lower, ranging from 73 to 95%. The two other cunner assemblies (pseudohaplotype GCA_020745675.1 and GCA_024362835.1) were incomplete in this region, underscoring the difficulty of assembling multigene families.
Fig. 3
Phylogenetic relationships of the GIMAP proteins found in the three wrasses via maximum-likelihood analysis of an alignment of the GTPase domains (Supplementary Fig. 1). The coloring of the labels matches the coloring of the genes in Fig. 2. The bootstrap values (%) are shown at each node. Note that cunner-a1 and -a4 are identical
AFP genes in the cunner are interspersed with GIMAP genes and share sequence similarityA total of seven proteins belonging to the GTPase IMAP family (GIMAPs) were encoded by genes interspersed among the AFP genes of the cunner (Fig. 2A). The ballan wrasse and spotty each had six GIMAPs (Fig. 2B-C), but AFP genes were not present at these loci or elsewhere in these genomes. The AFP genes share both proximity and sequence similarity with the GIMAP genes. The pair with the highest similarity was AFP11 with GIMAP-a5, where five segments had identities ranging from 73 to 98% (Fig. 2D). These segments lie both upstream and downstream of the coding sequence and overlap the first exon and the majority of the intron. The most notable difference between the loci is that the majority of the coding sequence within exon 2 is absent from the AFP. These similarities are sufficient to indicate that the AFP gene arose from a duplicated GIMAP-a gene.
Before the GIMAP genes were compared between the cunner, spotty and ballan wrasse, errors in the automated annotation of this repetitive gene family were corrected as described in the Materials and Methods. The accession numbers and sequences, if modified, are shown in Supplementary Table 1. Phylogenetic analysis indicated that the GIMAPs of these three species cluster into three groups, herein labeled type a, b or c (Fig. 3). Type c is restricted to spotty (four isoforms), where there is just one each of the type a and type b isoforms. Ballan wrasse and cunner have two divergently transcribed type b genes and four or five type a genes. These type a proteins cluster along species lines, with shorter branch lengths between the cunner isoforms, indicating that these genes were duplicated after the divergence of these two lineages and that this occurred more recently in the cunner. Taken together, these findings indicate that the GIMAP gene family is dynamic and that the AFP genes arose from a GIMAP-a gene, with subsequent tandem amplification of both genes within the cunner lineage.
Fig. 4
Sequences, models, and codon usage of cunner AFPs and Ala-rich C-terminal regions of GIMAP genes. A) AFP isoforms aligned with Ala highlighted in yellow, acidic and basic residues in red and blue font, respectively; Gly and Pro highlighted in pink, polar residues other than Thr highlighted in green, and aliphatic residues highlighted in gray, with the spacing of Thr residues (black highlighting) indicated above and asterisks indicating 100% conservation below. The last two residues (faded gray) are naturally removed when the C-terminus is amidated [29]. The AFPs are numbered sequentially as they appear in JAJGRF010000003.1, bases 6,121,000 to 6,371,000. B) Models of the long (AFP-1) and short (AFP-2) cunner AFPs generated using AlphaFold2-Colab [88] and rendered using PyMOL [87]. Residues are colored as above but with backbone atoms in light gray, Thr in green and other polar non-charged residues in dark green. Two 180°-degree rotations are displayed with their termini marked N and C. C) Ala-rich C-terminal regions of Cunner-A5 and Ballan-A1 GIMAPs relative to the shorter Cunner A isoforms. The Ala and Thr residues within the extension are colored as in A above, with the end of the GTPase domain in italics. Conservation between the unambiguously aligned residues of the four isoforms with extensions is indicated below the alignment by asterisks (all four sequences identical) or dots (three of four sequences identical). The beginnings of the two segments used to derive Ala codon usage are indicated with arrows. Accession numbers and reannotated sequences are given in Supplementary Table 1. D) Ala-codon usage of cunner AFPs (all isoforms, 322 codons) compared to the Ala-rich extensions of Cunner-A5 (26 codons), Ballan-A1 (75 codons), and Ala codons sampled from more than 5 million coding sequences from teleost fishes
The eleven cunner AFPs are highly similarThere are four AFP isoforms encoded by the eleven AFP genes (Fig. 4A), seven of which (2, 4–8, 10) match the previously characterized sequence [29]. Over 50% of the residues are Ala, and with one exception, Thr is spaced at 11-residue intervals. AFP9 differs from the main sequence at a single position (residue 4, Gly to Arg), while AFP11 contains one additional 11-a.a. repeat. Two genes (AFP1 and AFP3) encode identical isoforms that match AFP11, except that they contain an 18-a.a. insertion in which the Thr residues are spaced 18 residues apart.
Fig. 5
A comparison of snailfish AFP genes with respect to repeats, sequence identities, and codon biases. A) Schematics of an AFP-containing gene locus from dusky snailfish (GenBank accession JBEEID010000351 bases 1861 to 88,664) with the entire region, including the flanking genes ETV6 and PARP12, shown on top. An expansion of the AFP-containing region is shown beneath this in two segments. AFP coding sequences are indicated with blue arrows, repetitive elements with bars, and simple repeats with narrow red bars. Bars of the same color indicate that the repetitive elements are homologous and unique elements are shown in alternating shades of light and dark grey. The matching regions of AFP3 and the inverted AFP4 are indicated by black lines beneath. Segments corresponding to fragments that match the PARP12 gene are colored dark red and labelled. B) Characteristics of the locus encoding Tanaka-1 (GenBank accession JAYMGU010000011.1, 3811k-3878k), showing flanking genes (light brown). The expansion of the AFP-containing region is colored as above, with inverted repeats indicated with arrows and the matching region of the AFP and pseudogene by black lines beneath. C) Detailed schematics of four genes encoding AFPs from above. Repetitive elements identified as transposable elements (TEs) by Dfam [50] are indicated with wider bars and colored by type, other repetitive sequences are indicated by narrower bars with color indicating similarity. Matching segments are indicated with gray shading, with percent identity indicated. D) Ala codon usage in the AFPs from Fig. 6, the Ala-rich segment within intron 3 of PARP12 and a D. rerio Copia transposon
AlphaFold2 models of both the longest (AFP-1) and shortest (AFP-2) isoforms, in which the longer isoform has 29 additional residues, are very similar (Fig. 4B). Both form extended amphipathic α-helices that have a hydrophobic, Ala-rich surface punctuated by Thr residues. The other side of the helix is also enriched for Ala, but all of the charged residues are found here, many of which appear to form helix-stabilizing salt bridges. The disruption in the 11 aa spacing of the Thr residues by one 18 aa segment in the long isoform is an interesting deviation. An exact periodicity of 11 a.a. corresponds to 11 residues/3 turns, or 3.67 residues/turn, whereas the typical α-helix has 18 residues/5 turns, or 3.60 residues/turn. An examination of winter flounder AFP isoforms, including the crystal structure of a short isoform [34] and NMR structure of an engineered variant [55], as well as the crystal structure of the hyperactive isoform [35], revealed that residues at 11-a.a. intervals have a slight precession as the periodicity approaches ~ 3.65 residues/turn. Therefore, the 18 a.a. insertion serves to counteract this, bringing the Thr back into register (Fig. 4B, top).
Cunner AFP arose from the C-terminus of the GIMAP-a proteinThe Ala content of the GIMAP proteins is generally low. For example, cunner GIMAPa-1 has only seven Ala residues, making up 4% of the total. The four exceptions to this are cunner GIMAPa-5 (12%), ballan wrasse GIMAPa-1 (24%) and GIMAPa-4 (11%), plus spotty GIMAPa (10%). Their Ala-richness is restricted to the C-terminal region, which is outside of the GTPase domain, as shown in Fig. 4C. These extensions are present in all three wrasses being compared, whereas AFPs are found only in the cunner, so the Ala-rich extension arose prior to the AFP.
While these extensions are rich in Ala, they lack the periodicity of the Thr residues and contain more Gly and fewer charged residues than the AFPs. An AlphaFold2 model of the isoform with the longest C-terminal extension (Ballan-a1, Fig. 4C) predicts three α-helical segments in this region (Supplementary Fig. 2A), with the last spanning 37 aa (underlined in Fig. 4C) with 27 Ala residues (73%). The Ala-rich region of the shorter extension of Cunner-a5 is predicted to be unstructured (Supplementary Fig. 2B). Nevertheless, there is sufficient sequence similarity (Fig. 2D, darker yellow) to indicate that the Ala-rich extension gave rise to the AFP. The partial overlap of two of the matches is consistent with an internal duplication within the longer AFP11 allele. Interestingly, the similarity between the coding sequences was lower than that between the non-coding regions, consistent with positive selection of the AFP for its new function. Taken together, these data indicate that the AFP arose from a duplicated GIMAP-a gene containing an Ala-rich extension from which the GTPase domain was lost.
Ala codon usage of GIMAP-a and AFPs is similarly atypicalA further line of evidence that supports the Ala-rich extension of GIMAP-a as the progenitor of the AFP is that they share a similar codon usage bias. Cunner AFPs are unique among the type I AFPs in that Ala is preferentially encoded by GCT (72%), rarely by GCC (< 1%), and not at all by GCG (Fig. 4D). In contrast, in teleost fishes, GCT and GCC each encode approximately one-third of all Ala residues, while GCG encodes 11%. A similar bias is observed in the Ala-rich extensions, with Ballan-a1 employing GCT almost exclusively. This GCT bias is not observed in the flanking genes of any of these fishes (not shown), indicating that it is a characteristic of the C-terminal extension of the GIMAP-a genes that was retained in the AFP genes.
Part 2: SnailfishAFP sequences are only present in one genus of Liparidae (snailfishes)BLAST searches of genome sequences, the transcriptome shotgun assembly, and a selection of SRA datasets using both cDNA and protein sequences from snailfish AFPs [30, 56] revealed that in addition to species previously known to produce AFPs, namely, Atlantic (Liparis atlanticus), dusky (L. gibbus) and Tanaka’s (L. tanakae) snailfish, they are also found in L. liparis and L. tunicatus. Similar searches failed to identify homologs in other members of the same family (Liparidae) in different genera (Supplementary Table 2). However, the low complexity of snailfish AFPs (55–61% Ala, encoded primarily by GCC) means that divergent AFP sequences are difficult to identify.
Snailfish AFPs are members of a multigene familyThere are genome assemblies for both Tanaka’s snailfish and the dusky snailfish that were generated from long-read sequences. The dusky contig-level assembly was generated from PacBio sequences [49] and the Tanaka chromosome-level assembly from Oxford Nanopore sequences [48]. A dusky locus containing three AFP genes and a putative pseudogene is shown in Fig. 5A. Three AFP loci were previously identified from a short-read Tanaka genome assembly from a fish isolated from the Sea of Japan [56], but only one, located on chromosome 11, was found in the long-read genome assembly of the fish from the Yellow Sea (Fig. 5B) [49].
Fig. 6
Alignment of snailfish AFP sequences colored and annotated as in Fig. 4A. The GenBank accession numbers of the DNA sequences encoding these isoforms are Atlantic from cDNA; AY455862 [30]; Dusky-1 from a transcriptome; MT678484 [56]; Dusky-2 from cDNA; AY455863 [30]; Dusky-3 to 5 from genomic DNA, JBEEID010000351.1 bases 34,522 to 34,758, 42,139 to 41,882, 75,299 to 75,556 [49]; Tanaka-1 from chromosome 11, JAYMGU010000011.1 bases 3,855,161 to 3,855,574 [48]
Snailfish AFPs vary in length and sequence and largely lack regular Thr periodicityAn alignment of seven AFP sequences revealed three size classes, ranging from 78 to 85 a.a., 113 to 116 a.a, and 137 a.a. (Fig. 6). The Atlantic and Dusky-2 sequences are almost identical, with four differences restricted to the C-terminus. The three dusky sequences from the same locus (dusky-3, -4, and − 5, Fig. 6A) share 85 to 91% identity between themselves, whereas all other pairings drop below 70% identity.
Like all type I AFPs, the Ala content of these snailfish isoforms is high, ranging from 54 to 61%. However, the 11-aa Thr periodicity, which is prevalent in the cunner AFPs (Fig. 4A), is largely lacking, with each sequence having only one or two pairs of Thr residues with this spacing (Fig. 6). Nevertheless, Thr was the second most abundant residue in all of these sequences, ranging from 8 to 14%. Another notable difference is that the snailfish sequences all have two or more helix-breaking residues (Gly or Pro) around their midpoints (Fig. 6, pink highlighting) that are lacking in cunner AFPs (Fig. 4A).
The Tanaka AFP locus is absent from the dusky and hadal snailfishesThe single locus in the long-read Tanaka assembly contains one AFP gene and one AFP pseudogene (Fig. 5B, Supplementary Fig. 3A). This pseudogene shares 92% DNA sequence identity with Tanaka-1 but has two single nucleotide deletions (not shown) that disrupt the open-reading frame. These genes lie between the tensin-3 like protein (TNS3) and two convergently-transcribed isoforms of the insulin-like growth factor-binding protein (IGFBP) (Fig. 5B).
This locus was compared to the corresponding genomic region of a fish from the same family, the hadal snailfish, Pseudoliparis swirei [48]. These deep-water fish are unlikely to require an AFP as ice does not form at the constant near zero temperatures and high pressures found in the deep ocean [57]. As expected, type I AFPs were absent at this location, nor were they found elsewhere in the genome. A similar comparison was made with the Tanaka genome and again, AFP sequences were absent at this location. The genomic sequences of all three fishes aligned very well throughout most of their length in the flanking regions overlapping the TNS3 and IGFBP genes, indicated by the green lines on Fig. 5B, albeit with some insertions and deletions ≤ 1 kb in length. However, they did not share similarity to the Tanaka sequence in the the region between these genes, indicating that Tanaka’s snailfish was the only species of the three with AFP genes at this locus.
The dusky AFP locus is absent from the hadal and Tanaka snailfishesThere are three AFP genes and a putative AFP pseudogene found between the ETV6 and Poly(ADP-Ribose) Polymerase Family Member 12 (PARP12) genes of the dusky snailfish (Fig. 5A). The corresponding Tanaka locus is devoid of AFPs and similar to above, only matches the flanking genes as indicated by the green lines. The match with the hadal snailfish locus ended at the same spot near PARP12 but did not extend past the end of ETV6. The putative pseudogene (Supplementary Fig. 3B) may actually be a functional AFP, as although only the last third of the open reading frame resembles the other AFPs, the first two-thirds is Ala-rich. A monomeric model of this sequence suggests it could form a bundle of six helices of equal length (Supplementary Fig. 3C).
The coding sequences of Tanaka and dusky snailfishes are flanked by numerous TEsThe intergenic regions of both the dusky and Tanaka locus are populated largely by repetitive sequences, shown by bars in Fig. 5A, B. Many of these show similarity to transposable elements (TEs) detected by Dfam searches [50] and these are shown as wider bars in Fig. 5C and are listed in Supplementary Table 3. Additional segments (thinner bars) showed more than 80% identity to sequences present at least 100 times in several different fish genomes, suggesting that they are likely TEs that are not yet present in the Dfam database.
All of the AFP sequences are flanked by the same four TEs/repetitive elements (Fig. 5). The first flanking TE corresponds to the end of TE2 (magenta bars) and lies 23 bases upstream of each AFP gene and pseudogene. This segment was predicted to contain both a high-scoring TATA box (0.97) and initiation motif (0.84), with 16 bp of intervening sequence. This suggests that this portion of this TE, identified in an African cichlid (Supplementary Table 3), has been co-opted as a promoter. Additional segments overlapping a second region of this TE lie nearby or adjacent (dark purple bars).
The three additional flanking repetitive elements lie downstream of the genes. The first two (bright green and forest green bars) were not identified by Dfam. These are followed by TE3 (yellow). Even though these repetitive elements are shared, there are numerous insertions and deletions both within these elements and elsewhere that break up the matches. For example, Tanaka-1 contains an insertion of two segments of TE8 (light pink bars) between the coding sequence and the green elements, and a large portion of TE3 (yellow bars) was lost in dusky-3. Dusky AFP3 and AFP4 appear to be inverted repeats with some additional similarity (Fig. 5A) and dusky AFP 3 is flanked by inverted repeats of TE2 and TE5 (Fig. 5C), which suggests that these elements may have led to duplication of this gene. The regions between the AFPs share minimal similarity, as indicated by the alternating light and dark grey bars which correspond to repetitive sequences found only once at these loci. Those shown in other colors occur more than once. The majority of the low complexity sequence indicated by narrow red bars consists of dinucleotide repeats.
The snailfish AFP likely arose from a combination of repetitive non-coding DNA and transposonsGiven that the flanking sequences of the flounder [40], cunner (Fig. 2), and sculpin genes [58] were clearly associated with progenitor genes, it was presumed that the same would be true for snailfish. This may well be the case, although rather than being associated with any one gene, they are associated with a variety of TEs and putative TEs, suggesting that these could have been the progenitors (Fig. 5). Additionally, there are many instances of simple repeats within liparid genomes that have the potential to encode runs of Ala residues. One of these is found nearby, within intron 3 of PARP12 (Fig. 5, Supplementary Fig. 4A) and the DNA sequence can be aligned to that of dusky AFP3 with 67% identity (Supplementary Fig. 5A). Examples from other fishes include the Dada transposon from the Siamese fighting fish (Betta splendens) [59] that contains several stretches with Ala coding potential, the longest of which is 177 bp (not shown), or the Copia transposon from Danio rerio (Supplementary Fig. 4B) that can be aligned to dusky AFP3 with 62% identity (Supplementary Fig. 5B). An extreme example is (GCC)151, which is located within the last intron of the gene encoding potassium voltage-gated channel subfamily H member 5 (GenBank: XP_048106505) from Allis shad, which can be aligned to dusky AFP3 with 68% identity (Supplementary Fig. 5C). An origin from any one of these sequences could explain the biased codon usage (Fig. 5D) in which 75% of Ala codons are GCC, in contrast with the cunner AFP, where GCT is dominant (Fig. 4C).
Fragments of the PARP12 gene suggest the snailfish AFP gene arose in its vicinityThe sequence with Ala-coding potential in intron 3 of PARP12 is suggestive of a possible origin for the AFP gene, but as demonstrated above, the AFP coding sequence shares no more similarity to the AFP coding sequence than to (GCC)n. Therefore, the flanking regions were compared. No similarity was found to dusky-5 (Fig. 5C), or to any of the other AFP genes (not shown) within a 4 kb span that included the coding region.
However, beyond this span, there is evidence that small portions of the PARP12 gene were duplicated along with the AFP genes (Fig. 5A, B, dark red bars with labels). For example, a 322 bp segment found 2.9 kb upstream of dusky AFP4 matches a segment overlapping part of exon 1 and intron 1 of PARP12 with 97% identity (Supplementary Fig. 6A). Additional matches of 88% near Tanaka AFP6 and upstream of the putative dusky pseudogene are also found (Supplementary Fig. 6B, C). Therefore, it is possible that the N-terminal region of PARP12 spanning the region from exon 1 through intron 3 was duplicated, after which all but the segment with Ala-coding potential and small fragments were replaced by TEs, one of which provided a promoter for the nascent AFP gene. The sense orientation of the PARP12 fragments upstream of both dusky-4 and the putative dusky pseudogene support this hypothesis. However, the lack of clear homology adjacent to the AFPs could also suggest that the AFP gene arose entirely from repetitive elements in the vicinity of PARP12, in which a small portion of the 5ʹ region of the gene had been duplicated.
The AFPs of snailfish form a folded helix and may be dimericSnailfish AFPs are known to be largely helical [60], but there are two short regions where this is likely not the case. At the N-terminus, one or two Pro residues may prevent this segment from forming a helix (Fig. 6). The rest of the protein is roughly bisected by two helix-breaking residues (pink highlighting) spaced three to five residues apart. This is reminiscent of the large isoform of winter flounder, where a 195-a.a. alpha helix folds in half and then associates with another molecule as an antiparallel dimer that forms a four-helix bundle [35]. Therefore, the snailfish AFP was modeled here both as a monomer and a dimer.
The models that were generated (Fig. 7), whether for the monomer or the dimer, folded the polypeptide chain in the same manner. The first five residues were not predicted to form part of the helix. The rest of the chain was helical, with the exception of the bend, punctuated by Pro and/or Gly residues (pink). The helical segments on either side of the bend are predicted to lie alongside each other. In the dimeric model, the two monomers are antiparallel. Interestingly, the surface of one side of these models is very flat (Fig. 7B), and like other type I AFPs, it is dominated by Ala and Thr and devoid of charged residues, unlike the opposite surface (Fig. 7C). The dimer also appears plausible because there are several potential intermolecular salt bridges predicted from the antiparallel pairing (red and blue).
Fig. 7
Models of the dusky-1 AFP dimer (left) and monomer (right) generated using AlphaFold2-Colab [88] and rendered using PyMOL [87]. A) Cartoon representation of helices with the N (back) and C (front) termini indicated. B) Space filling model with the putative ice-binding surface facing forward. Residues are colored as in Fig. 4 but with backbone atoms in light grey, Thr in green, and other polar residues in dark green. C) View of the reverse side relative to B
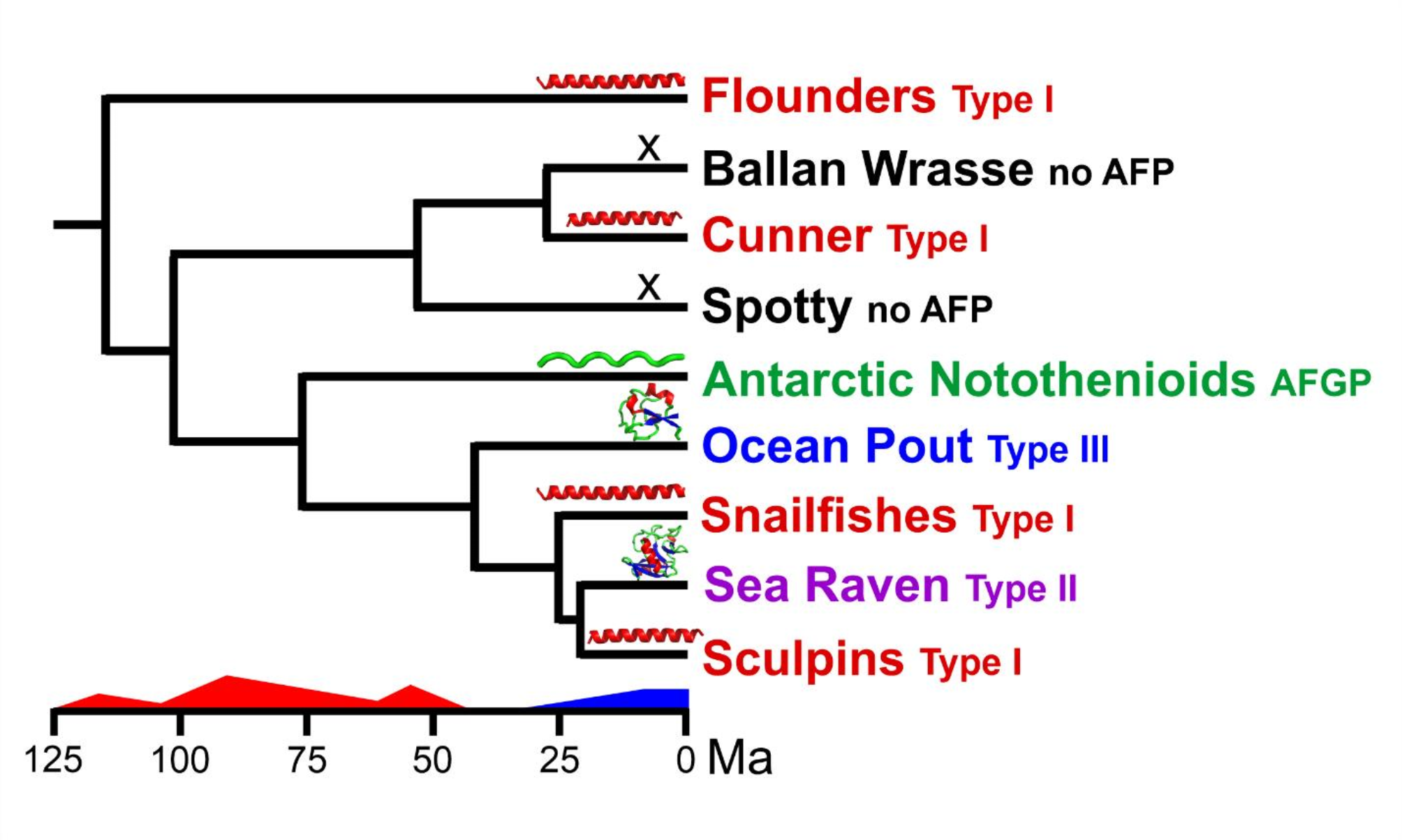
Part 3: Summary of the convergent origins of the four type I AFPsThe AFPs of flounders, sculpins, cunner and snailfishes arose recently enough, sometime within the last 30 Ma (Fig. 1), that the origins of all but snailfish AFP could be definitively traced to pre-existing functional genes via duplication and divergence due to the similarities that their non-coding regions have to other sequences. The cunner AFP arose from the GIMAP-a gene (Fig. 2), a conclusion that has been independently confirmed by Rives et al. [61], whereas the flounder AFP arose from a Gig-2 gene (Fig. 8A, B) [40], and the sculpin AFP arose from a lunapark gene (Fig. 8C) [58]. In the flounder, the antiviral Gig-2 genes were duplicated at a new location (not shown), and the AFP gene arose from a single copy of the preexisting Gig-2 gene (Fig. 8B). It was later duplicated at the site of origin multiple times, giving rise to a single locus containing multiple AFP genes in tandem (Fig. 8A), but Gig-2 genes were not retained at this location. In cunner (Fig. 2D), as in flounder (Fig. 8B), the gene structure and much of the flanking sequence of the progenitor were retained. However, in cunner, the GIMAP-a progenitor was retained at the site of origin of the AFP, as both genes were duplicated multiple times in situ (Fig. 2A). In sculpin, the 15-exon lunapark gene gave rise to the AFP (Fig. 8C), but only small portions of the original gene were retained in the AFP. Despite this, the AFPs of these three species share up to 80% identity, with similar N-termini (Met-Asp), Thr periodicity and Ala-richness, which would be indicative of homology in the absence of additional information (Fig. 8D). In contrast, the snailfish lacks short AFPs, and its longer isoforms display little Thr periodicity, but one isoform does begin with Met-Asp (Fig. 6). The snailfish AFP gene may have arisen from the PARP12 gene that is found flanking the dusky AFP region, but the sequence similarity is too limited to provide a definitive answer. What is evident is that the majority of the flanking sequence, and perhaps the coding sequence as well, likely arose from transposons and repetitive DNA (Fig. 5).
Fig. 8
留言 (0)