
記住我

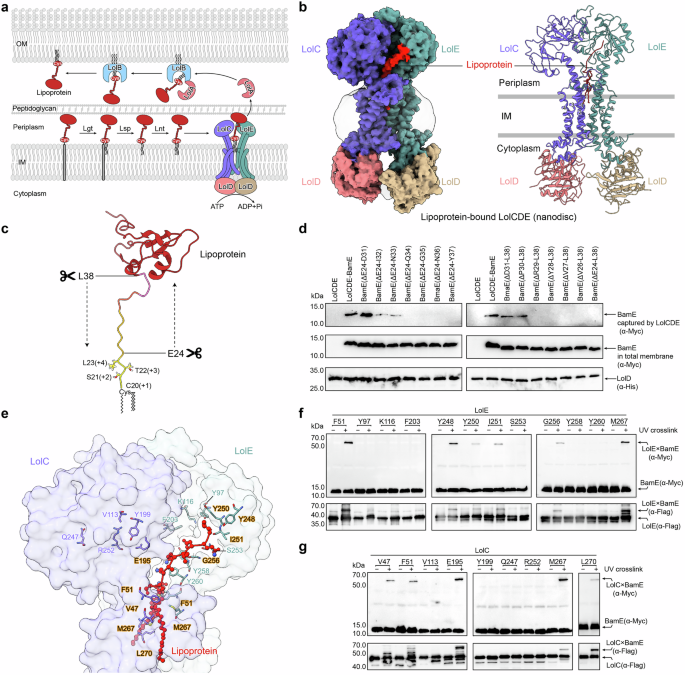



The cryo-EM structure of the lipoprotein-bound LolCDE complex reconstituted in nanodiscs was determined at a resolution of 3.5 Å, revealing a conformation highly consistent with the previously reported structure of lipoprotein-bound LolCDE in detergent (Fig. 1b and Supplementary Fig. 1). The lipoprotein is bound at the interface between the transmembrane helices (TMs) of LolC and LolE via its triacylated, unstructured N-terminal segment. Specifically, the triacyl chains and the initial (+1 to +4) residues of the lipoprotein are situated in the central cavity formed by the TM1 and TM2 of LolC and LolE, with the remaining segment extending outwards from the interface towards the periplasmic domain of LolE (Fig. 1b and Supplementary Fig. 2a). Notably, approximately half of OM lipoproteins possess a long, intrinsically disordered peptide linker at the N-terminus. This linker’s disordered nature is important for the proper relocalization of lipoproteins to the OM.33 To investigate whether the N-terminal linker affects the efficiency of lipoprotein binding by LolCDE, we systematically truncated the linker residues of the OM lipoprotein BamE from either end, while preserving the sorting signal residues (+1 to +4) (Fig. 1c). We then monitored the amount of BamE captured by LolCDE in vivo. Our findings indicate that at least ten residues of the N-terminal linker, including the triacylated cysteine, are necessary for efficient lipoprotein binding by LolCDE. Furthermore, the segment required for LolCDE binding appears to lack sequence specificity as truncations from either end yielded similar outcomes (Fig. 1d). Interestingly, this observation aligns with the length of the lipoprotein segment resolved in the LolCDE-lipoprotein structures, which spans from the central cavity to the periplasmic neck of TM2 in LolE (Supplementary Fig. 2b). The remaining structured body of lipoprotein has yet to be resolved in any reported LolCDE structure (Supplementary Fig. 2b), reflecting the flexibility of the unbound portion. These results indicate that LolCDE extracts lipoprotein requiring only the N-terminal triacyl chains and a small segment of the unstructured linker.
To identify LolCDE’s residues involved in lipoprotein binding, we carried out a UV-crosslinking assay on residues aligned within the path accommodating the triacyl chains and the linker in the LolCDE-lipoprotein structure (Fig. 1e). The assay revealed that residues F51 and M267 within the central cavity, G256 at the periplasmic neck of TM2, and Y248, Y250, and I251 further within the periplasmic domain of LolE effectively crosslinking with BamE (Fig. 1f). In contrast, within LolC, only the central cavity residues V47, F51, and M267, along with a periplasmic loop residue E195 near the TM2 neck of LolE, exhibited crosslinking with BamE (Fig. 1g). Notably, no crosslinking was observed at the periplasmic neck residues of TM2 in LolC. These results are consistent with prior binding analysis, which implicated LolE, but not LolC, in the interaction with lipoprotein.34 These results suggest that the triacyl chains of the lipoprotein interact with cavity residues in both LolE and LolC, whereas the lipoprotein linker is predominantly recognized by LolE via the periplasmic residues of TM2. This finding is consistent with the common feature observed in all reported lipoprotein-bound LolCDE structures, where the N-terminal linker of the lipoprotein is tilted towards LolE30,35,36 (Supplementary Fig. 2b).
The structural surface of the complex reveals highly hydrophobic patches surrounding the central cavity formed by TM1 and TM2, as well as the interface created by the gate loops extended between TM3 and TM4 (Supplementary Fig. 2c). Each of the triacyl chains (designated R1, R2, and R3) binds specifically within these hydrophobic pockets. The thioester-linked acyl chain, R1, is situated in the interface enclosed by the gate loop of LolC, while the amide-linked acyl chain, R3, and the other thioester-linked acyl chain, R2, are accommodated in a larger pocket enclosed by the gate loop (formerly known as the door bar) of LolE (Supplementary Fig. 2d). These specific interactions of the lipid chains and peptide linker to the complex indicate that the LolCDE extracts lipoprotein in a fixed orientation, allowing each structural element of the lipoprotein molecule to be precisely recognized by different domains of LolCDE.
LolC T302 and LolE T307 are involved in allosteric communications between the NBDs and TMDsOur previous finding revealed that the ATPase catalytic mutant LolCDE171QE reduced lipoprotein binding, whereas the ATP-binding mutant LolCDK48AE abolished lipoprotein transport in vitro30, implicating that lipoprotein transportation is triggered by ATP binding rather than hydrolysis. To investigate the molecular basis of ATP-bound LolCDE, we determined the structure of the mutant complex LolCDE171QE. The structure of E. coli LolCDE171QE reconstituted in nanodiscs was determined to 3.2 Å using cryo-EM (Fig. 2a and Supplementary Fig. 3). Despite the absence of added nucleotides, additional cryo-EM densities were observed in the catalytic pockets of LolD, which were identified as ATP molecules and magnesium ions (Fig. 2a and Supplementary Fig. 3). The overall conformation of the LolCDE171QE structure closely resembles the structures of LolCDE bound to ATP analogs, AMP-PNP and ADP-vanadate, with root-mean-square deviations (RMSD) of 1.2 Å over 405 aligned residues and 1.0 Å over 393 aligned residues, respectively30,35 (Supplementary Fig. 4a).
Fig. 2
Identification of the allosteric communicating residues of NBDs and TMDs of the LolCDE complex. a Cryo-EM density map (left) and cartoon representation (right) of LolCDE171QE structures in nanodiscs with ATP binding at 3.2 Å resolution. LolC and LolE are shown in purple and green, LolD dimer is shown in pink and yellow, and ATP is shown in gray, respectively. b Side and top views of the NBDs showing interactions with the coupling helices of LolC and LolE. The bound endogenic ATP molecules and magnesium ions are indicated. c Cell viability of the LolCDE mutants of the coupling helix interacting residues. Data are representative of n = 3 independent experiments. d Structural superimposition of the coupling helices of LolC (left) and LolE (right) and the NBDs of the lipoprotein-bound and ATP-bound LolCDE structures, showing distinct interactions at the coupling helices between the two structures
The nucleotide-binding domains (NBDs) of LolCDE171QE adopt a closed conformation, tightly sandwiching the endogenously captured ATP molecules between the RecA-like subdomain and the α-helical subdomain of opposing LolD subunits (Fig. 2b). Comparing the ATP-bound LolCDE171QE structure with the lipoprotein-bound structure, LolD dimerization induced remarkable conformational changes in the transmembrane domains (TMDs) and periplasmic domains (PDs) (Supplementary Fig. 4b). The NBDs interact with the TMDs through the coupling helices of LolC and LolE, which fit into the grooves between the RecA-like and α-helical subdomains of LolD (Fig. 2b). In the ATP-bound LolCDE171QE structure, a pair of polar residues, Q301 and T302, from the coupling helix of LolC interact with R85 on the side helix (S78-Q87) and Y93 on the Q-loop (I92-A104) of one LolD subunit, while R306 and T307 from the coupling helix of LolE interact with S78 on the side helix and Y93 on the Q-loop of the other LolD, respectively (Fig. 2b). To assess the importance of these polar residues to LolCDE’s function, we substituted them with alanine and conducted cellular viability assays (Fig. 2c). Since lipoprotein transport by LolCDE is an essential process in bacteria, disrupting the function of LolCDE leads to cellular lethality. The mutagenic assays were carried out using our previously reported lolCDE null strain HD200313, in which the expression of the lolCDE is controlled by the araBAD promotor.30 The results showed that the single mutant T302A in LolC, T307A in LolE, or Y93A in LolD, which are involved in the interactions between the coupling helix of LolC or LolE and the Q-loop of LolD, impaired bacterial viability (Fig. 2c and Supplementary Fig. 4c). These mutations did not affect the expression of the LolCDE complex (Supplementary Fig. 4d). Interestingly, these interactions were absent in the lipoprotein-bound structure, where the NBDs were in an undimerized conformation (Fig. 2d). This finding suggests that these interactions are crucial for coupling the conformational changes between the NBDs and TMDs in response to ATP binding.
LolC W249 and LolE W254 couple the conformational changes at the TMD-PD interfaceWhen comparing the TMDs of the ATP-bound and the lipoprotein-bound structures, the most striking conformational changes are observed in the long transmembrane helices TM1 and TM2 (Fig. 3a). In the ATP-bound structure, TM1 and TM2 of both LolC and LolE are more parallel, creating a closed interface as opposed to the V-shaped cavity observed in the lipoprotein-bound structure (Fig. 3a). This closed interface between LolE and LolC is incompatible with lipoprotein binding, as the TM2 helix of LolE in the ATP-bound LolCDE171QE structure would clash with the triacyl chains present in the lipoprotein-bound structure (Supplementary Fig. 5a). These observations suggest that the ATP-bound LolCDE171QE structure represents a post-transport state, where the bound lipoprotein is expelled from the central cavity as a result of the cavity closure induced by ATP binding. In this structure, TM2 of LolE undergoes an inward and upward movement, resulting in an approximately 90-degree bend at the periplasmic neck, transitioning from a relatively straight conformation in the lipoprotein-bound structure to a bent conformation in the ATP-bound state (Fig. 3a). Conversely, TM2 in LolC already exhibits a bent conformation in the lipoprotein-bound structure, with the state transition causing only a shift (Fig. 3a). Intriguingly, the periplasmic residues along TM2 of LolE and LolC display distinctly different chemical properties: while LolC residues are predominantly charged (W249RDRKGE255), those in LolE are primarily hydrophobic (W254IGTYGY260) (Fig. 3b). Notably, both LolC and LolE contain a conserved tryptophan residue, LolCW249and LolEW254, symmetrically positioned at the bending point of the TM2 neck (Fig. 3b), establishing extensive intermolecular contacts with surrounding residues from TM1, TM2, and the PDs (Fig. 3c). In particular, the aromatic side chain of LolCW249 interacts with L60 of TM1, R252 and L256 of TM2, and V232 of the periplasmic loop inLolC (Fig. 3c). Similarly, the aromatic side chain of LolEW254 makes contacts with L60, V63, and H65 of TM1, Y262 of TM2 and V231 of the periplasmic loop in LolE (Fig. 3c). Similar interactions involving LolCW249 and LolEW254 are also observed in the lipoprotein-bound structure (Supplementary Fig. 5b); however, these contacts are absent in the apo LolCDE structure (PDB code: 7ARI).
Fig. 3
Conformational change of the TM2 periplasmic neck of LolC and LolE in lipoprotein- and ATP-bound structures. a Structural superimposition of the lipoprotein-bound and ATP-bound LolCDE structures reveals conformational changes between TM helices of LolC and LolE. b Residues of the TM2 periplasmic necks of LolC and LolE are indicated. c A pair of tryptophan at the bend of the TM2 periplasmic neck of LolC and LolE, showing multiple contacts with surrounding residues in LolC (left) and LolE (right). d Cell viability of the mutants shown in (b). e Detections of the lipoprotein BamE binding to the mutant complexes shown in (d) by western blotting. Data in d, e are representative of n = 3 independent experiments
To evaluate the importance of these periplasmic residues for LolCDE’s function, we modified their chemical properties through site-directed mutagenesis and assessed their effects on rescuing the LolCDE null strain (Fig. 3d). Among the mutations examined, the mutations W254D in LolE and W249D in LolC completely abolished bacterial viability (Fig. 3d and Supplementary Fig. 5c–e). Additionally, the purified mutant protein complexes of LolCDEW254D and LolCW249DE exhibited diminished lipoprotein binding (Fig. 3e). These suggest that LolCDEW254 and LolCW249E are critical for the lipoprotein extraction function of LolCDE. Previous studies have shown that lipoprotein extraction by LolCDE occurs following ATP hydrolysis,30,35,36 which resets the LolCDE complex back to its relaxed apo conformation and disrupts the surrounding membrane to facilitate lipoprotein entry into the cavity. Notably, LolEW254 and LolCW249 do not directly interact with the bound lipoprotein, suggesting that their stabilized conformation between the TMDs and PDs is essential for efficient lipoprotein extraction.
We also investigated the periplasmic residues, LolEI251 or LolEY258, which are located along TM2 and associated with the unstructured linker. The mutants LolEI251D or LolEY258D exhibited reduced bacterial viability, but the purified mutant proteins showed normal lipoprotein binding (Fig. 3d, e and supplementary Fig. 5d, e). The milder effects suggest that these residues are important, but not essential for the function of LolCDE. In contrast, mutations in the TM2 residues on LolC showed no noticeable effect either in vivo or in vitro (Fig. 3d, e and Supplementary Fig. 5c, e).
Lipoprotein exits from the central core of the PDs for exportationThe periplasmic domains (PDs) of the ATP-bound LolCDE171QE structure exhibit prominent conformational changes compared to the lipoprotein-bound structure (Fig. 4a and Supplementary Fig. 6a). Each PD comprises two subdomains, Sabre and Porter,30 which together form a central core (Supplementary Fig. 6b). The shifts of TM2 elevate the base of the PDs, causing the periplasmic cores of LolC and LolE to enclose a cavity with the interior hydrophobic β-sheets facing each other (Supplementary Fig. 6b). To investigate whether these β-sheets constitute the path for lipoprotein export, we disrupted their continuity by introducing disulfide bonds between two opposing Sabre β-strands in LolC and LolE at the center. We then performed an in vivo viability assay and an in vitro lipoprotein transport assay to evaluate the effects. The in vitro transport assays were conducted according to a previously established protocol, using purified LolCDE reconstituted in phospholipid liposomes mixed with LolA, ATP, and magnesium ions to facilitate lipoprotein transport in vitro.30 Structural and biochemical studies indicate that the purified wild-type LolCDE complex naturally binds a lipoprotein in the substrate cavity, with lipoprotein delivery to LolA occurring in the presence of ATP and MgCl2.30,37,38 Cysteine mutations were introduced at residues LolCV111C and LolEA112C, which are in close proximity (less than 4 Å), on the β strands of LolC and LolE, respectively (Fig. 4a). Electrophoresis confirmed that the purified LolCV111CDEA112C mutant forms a disulfide-bonded complex, which could be reduced in the presence of DTT (Supplementary Fig. 6c, d). The in vivo analysis showed that the LolCV111CDEA112C mutant significantly diminished bacterial viability compared to the unaffected single cysteine mutations at LolCV111C or LolEA112C (Fig. 4b and Supplementary Fig. 6e). In vitro transport studies demonstrated that the crosslinked LolCV111CDEA112C complex reconstituted in phospholipid liposomes completely abolished lipoprotein transport to LolA when compared to the wild-type (Fig. 4c and Supplementary Fig. 6c, d). These results suggest that lipoprotein must traverse the central hydrophobic space enclosed by the β-sheets of the Sabre subdomains during its export to LolA.
Fig. 4
Analysis of lipoprotein export path between the periplasmic domains of LolCDE. a The hydrophobic cavity formed by the periplasmic cores of LolC and LolE in the ATP-bound structure of LolCDE. The hairpin loop of LolCE and the hook of LolE are highlighted with the indication of the residues mutated for the disulfide-bonded crosslink. b Cell viability of the mutants shown in (a). c In vitro lipoprotein transport assay of the disulfide-bonded mutants LolCV111CDEA112C and LolCA106CDEH177C to LolA. Data in b, c are representative of n = 3 independent experiments. d Conformational changes of the hooks of LolC and LolE in the lipoprotein-bound and ATP-bound structures
Furthermore, disrupting the integrity of this hydrophobic path by removing one side of the β strands proved detrimental (Fig. 4a, b). Deletion of the hairpin T101–Q114, which contains the interacting portion of the β strands from the Sabre subdomain of LolE, resulted in complete lethality without affecting the expression of the protein complex (Fig. 4a, b and Supplementary Fig. 6e, f). Notably, substituting the hydrophobic residue L103 or I113 with the charged residue aspartic acid within the Sabre β-strand of LolE was sufficient to cause lethality, whereas substituting the nearby polar residue T101 or Q114 by alanine had no effect (Fig. 4b and Supplementary Fig. 6e, f). Similarly, reversing the charges of E105 or R111 in the Sabre β-sheet of LolE had no effect (Fig. 4b and Supplementary Fig. 6e, f). These findings confirm that the hydrophobic pathway formed by the β-sheets of PDs is critical for LolCDE’s function, likely by accommodating the triacyl chains of the transported lipoprotein.
The PD of LolC associated with the periplasmic protein LolA via the hairpin hook structure for lipoprotein delivery.29 In the ATP-bound structure, the hook of LolC lifts and shifts towards the interface between the PDs, positioning itself to be accessible by both LolA and the bound lipoprotein (Fig. 4d). For lipoprotein transport to occur, the lipoprotein bound to the PD of LolE needs to ultimately reach the PD of LolC. However, the hook of LolE appears to obstruct the interface between the PDs of LolC and LolE, as it is situated close to an opposite periplasmic loop of LolC (Fig. 4d). This observation raises the question of whether an unimpeded interface between the PDs is essential for lipoprotein export. To test it, we crosslinked the proximate loops by introducing a disulfide bond through cysteine mutations at the LolE hook residue H177C and the LolC loop residue A106C (Fig. 4a). The purified LolCA106CDEH177C protein forms a disulfide-bonded complex that is reducible by DTT (Supplementary Fig. 6d, g). The LolCA106CDEH177C mutant was lethal in vivo and inhibited lipoprotein transport to LolA in vitro (Fig. 4b, c and Supplementary Fig. 6d, e, and g). These results suggest that the delivery of lipoprotein to LolA necessitates passage through the interface between the PDs. The flat conformation of the LolE hook observed in the ATP-bound structure may play a role in preventing lipoprotein backflow. Additionally, deletion of the LolE hook segment P171–P182 also resulted in lethality (Fig. 4b and Supplementary Fig. 6e). However, the expression of the LolCDE∆P171-P182 mutant protein was markedly reduced (Supplementary Fig. 6f), indicating that the hook of LolE is crucial for structural stability of the complex.
Identification of lipoprotein binding residues in the periplasmic core of LolCDEThe conformational changes in the periplasmic necks of transmembrane helices caused the PDs to move closer together, with the PD of LolE exhibiting the most pronounced alternations (Supplementary Fig. 4b and Supplementary Fig. 6a). In the ATP-bound structure, the helmet-shaped PDs of LolC and LolE come together to create a periplasmic cavity. Both the PDs of LolC and LolE contain a hydrophobic core, which connects with the hydrophobic cavity of the TMDs (Supplementary Figs. 2d and 7a). To determine whether the residues in the hydrophobic core are essential for the functionality of LolCDE (Fig. 5a), we performed single-site mutagenesis and assessed cellular viabilities. Substituting the residues L60, V63, and H65 in the periplasmic neck of TM1 in LolE with aspartic acid (L60D, V63D, and H65D) led to lethality or severe viability defects (Fig. 5a, b and Supplementary Fig. 7b). Additionally, mutations L199D and F203D from the Sabre subdomain, as well as Y97D and L126D in the region between the Sabre and Porter subdomains of LolE, also resulted in bacterial death or viability defects (Fig. 5b and Supplementary Fig. 7b). Western blot analysis confirmed that the expression levels of the mutant proteins were not affected (Supplementary Fig. 7c), suggesting that the inhibitory effects were due to functional defects rather than protein expression defects. These results indicate that the internal residues of the periplasmic core of LolE are crucial for the function of LolCDE. In contrast, the mutants in the periplasmic residues of LolC had milder effects. Only the L60D and M63D mutations in TM1 of LolC caused severe viability defects (Fig. 5b and Supplementary Fig. 7b, d), whereas the V196D and Y199D mutations within the Sabre subdomain, and T98D in the region between the Sabre and Porter subdomains, had minimal impact on bacterial viability (Fig. 5b and Supplementary Fig. 7b, d). Interestingly, the L60D and M63D mutants in LolC also showed abolished lipoprotein binding (Fig. 5c), whereas lethal mutants in LolE did not affect lipoprotein binding (Fig. 5d). L60 and M63 are located on the TM1 periplasmic neck of LolC and interact with W249 on TM2 (Fig. 3c). Disrupting the hydrophobicity of L60, M63, and W249 consistently led to lethality and loss of lipoprotein binding (Figs. 3c and 5c), suggesting that the periplasmic conformation of LolC stabilized by these hydrophobic interactions is essential for lipoprotein extraction. In contrast, mutations at L60, V63, and H65, which interact with W254 of LolE at the periplasmic neck, also caused lethality but maintained relatively normal lipoprotein binding levels. This suggests that these hydrophobic residues of LolE have functions beyond lipoprotein binding, likely contributing to the stabilization of the conformational changes of the PD required for lipoprotein transport.
Fig. 5
Functional analysis of the periplasmic central core of LolCDE. a Cartoon representation of the periplasmic domains of LolE (left) and LolC (right) showing the hydrophobic residues at the periplasmic central cores. b Cell viability of the mutants shown in (a). c, d Lipoprotein binding analysis in the mutant complexes shown in (b) by western blot. Data in b, c are representative of n = 3 independent experiments
留言 (0)