
記住我

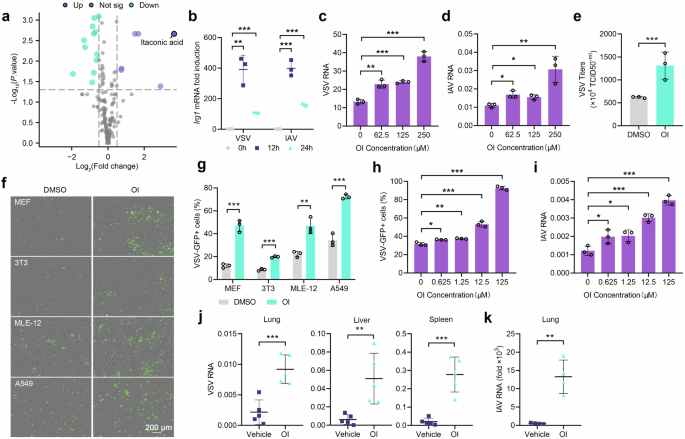



To investigate the metabolic alterations following viral infection, metabolomics analysis was performed in mouse lung 12 h after intraperitoneal (i.p.) infection with vesicular stomatitis virus (VSV). The results revealed itaconate as the predominant elevated metabolite post infection (Fig. 1a). Meanwhile, the expression of itaconate synthetase IRG1 in lung, liver, and spleen was significantly increased at 12 h after i.p. infection with VSV or herpes simplex virus type I (HSV-1) (Supplementary Fig. 1a, b). Additionally, nasal infection with VSV and influenza A virus PR8 (IAV) also led to a significant induction of Irg1 expression at 12 h (Fig. 1b).
Fig. 1
Inducible IRG1-itaconate axis facilitates viral infection. a Metabolites in the lungs from mice i.p. infected with VSV or not (n = 6). b Irg1 mRNA expression in the lungs of mice intranasally (i.n.) infected with VSV and IAV (n = 3). c, d Viral RNA in PMs pretreated with different concentrations of OI, infected with VSV (c) or IAV (d) (n = 3). e Viral titers in the supernatant of PMs pretreated with OI (250 μM) or DMSO (n = 3). f, g VSV-GFP infection in MEF, 3T3, MLE-12 and A549 cells pretreated with OI (125 μM) or DMSO, detected by fluorescence (f) and flow cytometry (g) (n = 3). h, i VSV-GFP (h) and IAV (i) infection in MLE-12 cells pretreated with different concentrations of OI. j, k Mice were pretreated with OI, subsequently infected with VSV (i.p.) and IAV (i.n.), and viral RNA loads were detected (n = 5 or 4). Data are mean ± SD or representative of 3 independent experiments with similar results. *p < 0.05, **p < 0.01, ***p < 0.001 by an unpaired, two-tailed t-test
Next, we investigated the biological function of itaconate in viral infection. We employed a cell-permeable derivative of itaconate, 4-octyl itaconate (OI), which closely mimics the biological functions of itaconate.19 Treatment of peritoneal macrophages (PMs) with OI increased intracellular VSV, IAV (Fig. 1c, d) and HSV-1 (Supplementary Fig. 1c) genome load. Correspondingly, viral titers were confirmed to be higher in culture medium with OI treatment (Fig. 1e and Supplementary Fig. 1d). Similar results were observed in PMs treated with underivatized itaconate as well (Supplementary Fig. 1e–g). Besides macrophages, viruses also infect epithelial cells, endothelial cells and fibroblast cells.20 Consistently, OI also enhanced the infection in pulmonary epithelial and fibroblast cell lines including MEF, 3T3, MLE-12 and A549 cells (Fig. 1f, g). Additionally, an OI concentration as low as 0.625 μM was sufficient to promote viral replication in MLE-12 cells (Fig. h, i). We further investigated the in vivo effect of itaconate, and pre-treatment with OI significantly increased their susceptibility to VSV and IAV infections (Fig. 1j, k). These findings indicate that itaconate and OI could promote various viral infections.
Itaconate and its derivatives have been reported to regulate type I interferon (IFN-I) production.6,19 To determine whether the effect of itaconate on viral infection was attributed to IFN-I pathway, we explored the effect of itaconate in Irf3-/- and Ifnar1-/- macrophages. Despite the deficiency of IFN-I pathway in these cells, OI still significantly augmented VSV and IAV infection (Supplementary Fig. 2a-e). Similarly, blockade of interferon receptor with blocking antibody failed to abrogate the enhancing effect of OI on VSV and IAV infection (Supplementary Fig. 2f, g). Conversely, OI failed to promote HSV-1 infection in macrophages deficient in IFN-I pathway, indicating its reliance on IFN-I (Supplementary Fig. 2h, i). This is consistent with the previous findings that itaconate exert inhibitory effects on IFN-I production through itaconation of STING.21,22 Taken together, these data indicate that OI control VSV and IAV infection independent of IFN-I signaling.
IRG1 facilitates VSV and IAV infection through itaconateWe subsequently investigate the impact of endogenous IRG1 on viral infection. Irg1 exhibited significant upregulation among metabolic genes upon viral infection (Fig. 2a, b). Viral infection induced significant IRG1 expression (Fig. 2c) and itaconate production (Fig. 2d) in macrophages. Functionally, Irg1 deficiency attenuated VSV and IAV infection (Fig. 2e, f), resulting in a decreased IFN-I production, ISGs expression, including ubiquitin-like protein ISG15 (Isg15) and interferon-induced GTP-binding protein Mx1 (Mx1), and TBK1-IRF3 signaling pathway activation (Fig. 2g–k). To further elucidate the role of IRG1 in the immune response to cytoplasmic RNA, we stimulated macrophages with ultraviolet irradiated VSV (UV-VSV) and intracellular transfection of poly I:C. We observed that knockout of IRG1 slightly attenuated the immune responses to UV-VSV and poly I:C (Fig. 2l, m). However, viral infection was paradoxically inhibited in Irg1-/- macrophages (Fig. 2e, f), indicating the enhancing effect of IRG1 on RNA virus replication surpasses its impact on innate immune response. Moreover, Irg1 silencing still impeded VSV infection in Irf3-/- macrophages (Fig. 2n and Supplementary Fig. 2j, k). Taken together, these data indicate that endogenous IRG1 improves the susceptibility of macrophages to VSV and IAV infection through an IFN-I independent manner. In contrast, Irg1 loss reduced HSV-1 infection while increased the production of IFN-I (Fig. 2o, p), which can be attributed to its negative regulation of the STING-mediated immune response.23
Fig. 2
IRG1 facilitates VSV and IAV infection independent of IFN-I pathway. a The most significantly altered metabolic genes upon VSV infection were identified by RNA-seq (n = 2). b Irg1 mRNA expression in PMs infected with VSV, IAV or HSV-1 for 8 h (n = 3). c Immunoblot of IRG1 in PMs infected with VSV or IAV. d Itaconate in the supernatants of PMs infected with VSV for 12 h (n = 3). e, f Viral RNA in Irg1+/+ and Irg1-/- BMDMs infected with VSV (e) or IAV (f) (n = 3). g ELISA analysis of IFN-β in the supernatants of Irg1+/+ and Irg1-/- BMDMs infected with VSV for 24 h (n = 3). h Isg15 and Mx1 mRNA expression in Irg1+/+ and Irg1-/- BMDMs infected with VSV (n = 3). i Immunoblot of IRG1, RIG-I, MAVS, TBK1, phosphorylated TBK1 (p-TBK1), IRF3, phosphorylated IRF3 (p-IRF3), STAT1, phosphorylated STAT1 (p-STAT1) in Irg1+/+ and Irg1-/- BMDMs infected with VSV. j, k Ifnb1 (j) and Isg15 (k) mRNA expression in Irg1+/+ and Irg1-/- BMDMs infected with IAV (n = 3). l Ifnb1 mRNA expression in Irg1+/+ and Irg1-/- BMDMs transfected with poly I:C (3 ug/ml) for 8 h (n = 3). m Ifnb1 and Isg15 mRNA expression in Irg1+/+ and Irg1-/- BMDMs infected with UV-VSV (n = 3). n VSV RNA in Irf3-/- PMs carrying Irg1 or control siRNA (n = 3). o HSV-1 RNA in Irg1+/+ and Irg1-/- BMDMs (n = 3). p ELISA analysis of IFN-β in the supernatants of Irg1+/+ and Irg1-/- BMDMs infected with HSV-1 for 24 h (n = 3). q VSV RNA in Irg1+/+ and Irg1-/- BMDMs pretreated with OI (250 μM) or DMSO (n = 3). r The effect of OI on VSV-GFP infection rate in Irf3-/- PMs carrying Irg1 or control siRNA (n = 3). Data are mean ± SD or representative of 3 independent experiments with similar results. *p < 0.05, **p < 0.01, ***p < 0.001, N.S, not significant by an unpaired, two-tailed t-test
Considering that endogenous IRG1 can function in an itaconate-independent manner,24,25 we treated Irg1-/- macrophages with OI following viral infection. The supplementation of OI to Irg1-/- macrophages greatly enhanced VSV infection and rescued the impaired viral infection caused by Irg1 deficiency (Fig. 2q), even in an IFN-I-deficient situation (Fig. 2r). These data suggest that the IRG1-itaconate axis promotes viral infection independent of IFN-I.
Antioxidant alterations is not responsible for OI-mediated facilitation of viral infectionWe next investigated the mechanisms underlying the promotion of VSV and IAV infection by itaconate. The biological functions of the IRG1-itaconate axis are suggested to rely on its regulation of antioxidant response.26 Indeed, OI suppressed ROS production in macrophages with VSV infection (Supplementary Fig. 3a, b). However, ROS scavengers, NAC (N-acetyl-L-cysteine) and Tempol, had no effect on OI-mediated enhancement of VSV infection (Supplementary Fig. 3c, d). Furthermore, Nrf2 is characterized as a sensor of oxidative stress and regulated by itaconate.6 Consistently, we found that OI significantly increased Nrf2 translocation into the nucleus upon VSV infection (Supplementary Fig. 3e). However, Nrf2 silencing did not affect OI-induced viral overgrowth (Supplementary Fig. 3f–h). Thus, the promotion of VSV infection by OI is not mediated through the regulation of antioxidant responses.
Glucose metabolism disturbance is not responsible for OI-mediated facilitation of viral infectionItaconate also exerts a regulatory effect on glycolysis and oxidative respiration by specifically targeting key enzymes such as GAPDH14,15 and SDH.27 Therefore, we investigated the role of glucose metabolism in facilitating viral infection via OI. ATP significantly enhanced viral replication (Supplementary Fig. 4a). However, the activation of IRG1-itaconate axis reduced both ECAR and OCR (Supplementary Fig. 4b, c), suggesting the negative regulation of glycolysis, oxidative respiration, and impaired ATP generation. Furthermore, ATP synthase inhibitor, oligomycin, significantly impeded viral replication, while the enhancing effect of OI on viral infection remained unaffected (Supplementary Fig. 4d).
A recent report revealed that neuronal itaconate inhibits the activity of SDH, leading to a metabolic state that suppresses Zika virus replication.27 To determine this in our situation, we pretreated macrophages with dimethyl malonate (DMM), a classical inhibitor of SDH, and found that DMM had no significant effect on VSV infection in vitro (Supplementary Fig. 4e-g). Furthermore, in vivo data revealed that neither viral burden nor cytokines production was affected in mice treated with DMM (Supplementary Fig. 4h, i). Taken together, these findings indicate that the regulation of glucose metabolism via the IRG1-itaconate axis is not a pivotal factor in promoting VSV infection.
Geranylgeranyl diphosphate is required for OI-mediated facilitation of viral infectionTo identify the mechanisms of the IRG1-itaconate activity during viral infection, we performed transcriptome RNA-sequencing analysis in Irg1+/+ and Irg1-/- macrophages. Differentially expressed genes are significantly enriched in the pathways related to the classical functions of itaconate, such as “response to oxidative stress” and “regulation of inflammatory response” (Fig. 3a). Additionally, we observed a significant enrichment of differentially expressed genes in the pathways associated with fatty acid metabolism and cholesterol metabolism (Fig. 3a), suggesting the potential involvement of the IRG1-itaconate axis in lipid metabolism. Consistently, OI treatment also induced a broad change in genes related to lipid metabolism-associated signaling pathways, especially cholesterol and fatty acid metabolic processes (Fig. 3b). Therefore, we next focused on the relationship between the IRG1-itaconate axis and lipid metabolism.
Fig. 3
GGPP is indispensable for facilitating viral infection induced by OI. a, b GO Pathway enrichment analysis of differentially expressed genes from the RNA-seq data of Irg1+/+ and Irg1-/- BMDMs (a), and PMs treated with OI (250 μM) or DMSO (b). c The effect of OI on VSV-GFP infection rate in MLE-12 cells pretreated with TOFA (30 μM) or vehicle (n = 3). d, e The effect of OI on VSV infection in MLE-12 cells (d) and PMs (e) pretreated with simvastatin (Sim, 10 μM) plus GGOH (15 μM), FOH (15 μM), SQE (15 μM) or not (n = 3). f Schematic of mevalonate pathway and protein prenylation. g The effect of OI on VSV-GFP infection rate in MLE-12 cells pretreated with simvastatin (Sim, 10 μM) with or without a cell permeable cholesterol (Chol, 5 ug/ml) (n = 3). Data are mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, N.S, not significant by an unpaired, two-tailed t-test
Firstly, we investigated the role of fatty acids and cholesterol synthesis in OI-mediated viral overgrowth. TOFA, an inhibitor of fatty acids synthesis, did not affect viral overgrowth caused by OI (Fig. 3c and Supplementary Fig. 5a). However, simvastatin and lovastatin, inhibitors of the rate-limiting enzyme HMG-CoA reductase (HMGCR) in cholesterol synthesis,28 strongly abrogated the viral overgrowth caused by OI (Fig. 3d, e and Supplementary Fig. 5b), indicating that OI-mediated viral overgrowth largely depends on some metabolic pathways downstream of HMGCR.
Inhibition of HMGCR is known to reduce the metabolic intermediate mevalonate, which ultimately results in a drop in cholesterol and other biosynthetic intermediate isoprenoids, including farnesyl diphosphate (FPP) and geranylgeranyl diphosphate (GGPP)29 (Fig. 3f). To determine the key intermediates, we analyzed whether OI affected cholesterol production and distribution. Neither Irg1 deficiency nor OI treatment changed total- or free-cholesterol levels in MLE-12 cells and macrophages (Supplementary Fig. 5c, d). The distribution of cholesterol remained unaffected upon treatment with OI (Supplementary Fig. 5e). In addition, supplementation of squalene (SQE) (Fig. 3d, e), the precursor of cholesterol, or a cell permeable form of cholesterol (Fig. 3g and Supplementary Fig. 5f) failed to rescue the activity of statins on OI-mediated viral overgrowth, indicating that the effect of OI is independent of cholesterol.
We next investigated the role of isoprenoids on viral infection induced by OI. In mevalonate pathway, mevalonate sequentially transforms to isopentenyl pyrophosphate (IPP), FPP and GGPP (Fig. 3f). Addition of permeable geranylgeraniol (GGOH), the donor of a geranylgeranyl group (Fig. 3f), almost fully rescued the inhibitory activity of simvastatin in both MLE-12 cells and macrophages (Fig. 3d, e). Conversely, farnesol (FOH), which acts as a donor of a farnesyl group and was unable to rescue GGPP synthesis in the absence of IPP28 (Fig. 3f), exhibited an inability to restore the observed OI effect in macrophages (Fig. 3e). Collectively, these data suggest that isoprenoids, especially GGPP, may be implicated in the impact of OI on viral infection.
Itaconate promotes the membrane localization of Rab GTPasesGGPP serves as the substrate for a distinct type of protein prenylation called geranylgeranylation (Fig. 3f). Protein prenylation is responsible for anchoring soluble proteins to cellular membranes.29 Geranylgeranylation is primarily catalyzed by two heterodimeric prenyltransferases, namely geranylgeranyltransferase type I (GGTase-I), and Rab geranylgeranyl transferase (GGTase-II, RGGT, RabGGTase)30 (Fig. 3f). These two prenyltransferases exhibit specificity towards certain target proteins.
To investigate whether OI affected the subcellular localization of the classical target proteins of GGTase-I and GGTase-II, the cytoplasmic and membrane proteins of MLE-12 cells treated with OI or vehicle were isolated, followed by immunoblot analysis. The result displayed that OI had no effect on the distribution of GGTase-I’s target proteins, including Rac, Rap and RhoA (Supplementary Fig. 6a). However, it significantly increased the membrane localization of Rabs (including Rab1a, Rab5, Rab6, Rab7 and Rab11), the classical targets of GGTase-II, and decreased their cytoplasmic localization without altering total protein levels (Fig. 4a). Rab5 and Rab7, which localized on early endosomes and late endosomes respectively, exhibited a greater tendency towards endosomal membrane localization upon OI treatment (Fig. 4b). Moreover, the regulatory impact of OI on Rabs’ distribution was abolished by 3-PEHPC, a GGTase-II inhibitor, or simvastatin that blocks GGPP production (Fig. 4c). These data suggest that OI enhances the subcellular distribution of Rabs on the membrane, while GGPP and GGTase-II-mediated geranylgeranylation on Rabs is necessary for this effect. Additionally, the promotion of VSV and IAV infection by OI was abolished by GGTase-II inhibitor 3-PEHPC, but not GGTase-I inhibitor GGTI-298 (Fig. 4d, e). A similar result was observed upon itaconate treatment (Supplementary Fig. 6b). Taken together, these data indicate that GGTase-II-mediated geranylgeranylation is required for regulating the localization of Rabs and enhancing viral infection by OI.
Fig. 4
OI induces the redistribution of Rab GTPases to enhance viral infection. a Immunoblot of GGTase-II substrate proteins in the membrane, cytoplasm and total cell fractions of MLE-12 cells pretreated with OI (125 μM) or DMSO. b Confocal of Rab5 and EEA1 co-localization (n = 20), Rab7 and LAMP1 co-localization (n = 23), in MLE-12 cells pretreated with OI or DMSO. The icon on the right displays the co-localization rate. c The effect of OI on Rab5 and Rab7 distribution in the membrane and cytoplasm in MLE-12 cells treated with simvastatin (Sim, 10 μM), 3-PEHPC (1.5 mM) or vehicle as indicated. d, e VSV-GFP (d) and IAV (e) infection in MLE-12 cells pretreated with OI or DMSO plus FTI-277 (10 μM), GGTI-298 (10 μM), 3-PEHPC (1.5 mM) or vehicle for 12 h (n = 3). f VSV RNA in PMs and MLE-12 cells pretreated with OI or DMSO 1 h post infection (n = 3). g VSV RNA in Irg1+/+ and Irg1-/- BMDMs infected with VSV for 1 h (n = 3). h, i VSV RNA in MLE-12 cells pretreated with OI or DMSO (h), and in Irg1+/+ and Irg1-/- BMDMs (i) infected with VSV for 1 h at 4 °C (n = 3). j, k VSV RNA in MEF cells pretreated with OI or DMSO (j), and in Irg1+/+ and Irg1-/- MEF cells (k) transfected with VSV-RNA (n = 3). l, m The effect of OI on VSV-GFP (l) and IAV (m) infection in MLE-12 cells silenced of Rab5, Rab7, or Rab11 (n = 3). Data are mean ± SD or representative of 3 independent experiments with similar results. *p < 0.05, **p < 0.01, ***p < 0.001, N.S, not significant by an unpaired, two-tailed t-test
In addition, we also investigated another type of protein prenylation known as farnesylation, which is mediated by the FPP-farnesyltransferase (FTase) axis (Fig. 3f). OI induced a redistribution of Ras, a classical target protein of FTase, between cytoplasm and membrane (Supplementary Fig. 6c). However, the enhancing effect of OI on viral infection remained unaffected by treatment with the FTase inhibitor FTI-277 (Fig. 4d, e). Thus, FTase is not responsible for the observed effect of OI on viral infection.
The IRG1-itaconate axis promotes viral entry and post-entry processesGiven that viruses exploit Rab GTPases, the master regulators of intracellular membrane trafficking, to facilitate their entry, uncoating and budding processes,16 we explored the specific life stages in which the IRG1-itaconate axis affects viral infection. Notably, OI increased VSV entry in a dose-dependent manner (Fig. 4f), while Irg1 deficiency impaired VSV entry in macrophages (Fig. 4g). Meanwhile, neither OI treatment nor Irg1 deficiency affected viral binding (Fig. 4h, i). Thus, the IRG1-itaconate axis acts on the early entry of VSV infection.
To assess the impact of the IRG1-itaconate axis on post-entry processes of viral infection, the cells were transfected with viral genomic RNA to bypass the binding and entry stages of VSV infection. VSV genome load was also elevated by OI (Fig. 4j). In accordance, viral genome load was significantly lower in Irg1-/- group (Fig. 4k). Collectively, these results reveal that the IRG1-itacoante axis promotes VSV infection via facilitating both viral entry and post-entry processes.
We next investigated the role of the crucial Rab GTPases associated with viral entry, uncoating and egress processes.16,31 Rab5, Rab7 or Rab11was silenced respectively following viral infection (Supplementary Fig. 7a-c). Indeed, silencing Rab5, Rab7 and Rab11 attenuated the increased VSV and IAV infections induced by OI (Fig. 4l, m). Therefore, the impact of OI on viral infection is dependent on various Rabs associated with viral entry and post-entry processes.
Itaconation of GDI2 impedes the extraction of Rab GTPases from the membraneSubsequently, we aim to elucidate the mechanism by which itaconate regulates the subcellular localization of Rab proteins. Once a Rab protein is synthesized, it is escorted by Rab escort protein (REP) to GGTase-II for geranylgeranylation, thereby acquiring the ability to bind to membranes. Subsequently, Rabs shuttle between the membrane and cytoplasm in a tightly regulated manner regulated by various proteins17 (Fig. 5a). We initially investigated whether OI affected the geranylgeranylation on Rab proteins using GGOH-azide, a probe for studying geranylgeranylation.32,33 OI treatment resulted in a decrease of the geranylgeranylation of Rabs, either overall or individually (Supplementary Fig. 8a–c), indicating that OI does not increase the membrane delivery of Rabs through up-regulating the geranylgeranylation of Rabs.
Fig. 5
OI inhibits the extraction of Rabs from the membrane via alkylation of GDI2. a Schematic of Rab GTPases cycle. b, c The effect of OI on VSV-GFP (b) and IAV (c) infection in MLE-12 cells silenced of GDI1 or GDI2 (n = 3). d Immunoblot of Rab5 and Rab7 in the membrane and cytoplasm of MLE-12 cells carrying GDI2 or control siRNA, and treated with OI (125 μM) or DMSO. e, f The effect of OI on the interaction of GDI2 (e) and GDI1 (f) with the Rabs in HEK293T cells transfected with GDI2-Myc or GDI1-Myc together with Flag-Rab5, Flag-Rab7 or Flag-Rab11. g Immunoblot of ITalk-modified (ITalked) GDI1 and GDI2 in HEK293T cells transfected with GDI1-Myc or GDI2-Myc. h Immunoblot of ITalked GDI2 in HEK293T cells treated with different concentrations of OI (0, 62.5 or 125 μM). i ITalked endogenous GDI2 proteins in MLE-12 cells treated with ITalk or DMSO. j LC-MS/MS analysis of itaconation on GDI2 in HEK293T cells transfected with GDI2-Flag, treated with OI for 12 h. k The effect of OI on IAV infection in GDI2-deficient MLE-12 cells transfected with wildtype or mutated GDI2 (n = 3). l Immunoblot of GDI2 in the lungs from mice intratracheally administered with adeno-associated virus containing GDI2 shRNA (shGDI2) or scrambled shRNA (shCtrl) (n = 3 per group). m VSV RNA in the lungs of mice upon GDI2 knockdown and control mice (n = 4). n IAV RNA in the lungs of mice upon GDI2 knockdown and control mice (n = 3). Data are mean ± SD or representative of 3 independent experiments with similar results. *p < 0.05, **p < 0.01, ***p < 0.001, N.S, not significant by an unpaired, two-tailed t-test
Increased membrane localization of Rabs can be attributed to an increased delivery to membrane or a reduced dissociation from it, we subsequently focused on the dissociation process of Rabs from the membrane. Rab GDP dissociation inhibitors (GDI1 and GDI2) bind to geranylgeranylation groups on the inactive form of Rabs and extract them from the membrane and enabling their availability for another round of membrane delivery18 (Fig. 5a). Dysfunction of GDIs leads to the retention of Rabs within the membrane. To determine whether OI-induced viral overgrowth was dependent on GDIs, we respectively silenced GDI1 and GDI2 (Supplementary Fig. 9a, b). Notably, silencing GDI2 but not GDI1 abolished the promotion of VSV and IAV infection caused by OI (Fig. 5b, c). Moreover, silencing GDI2 significantly impeded the dissociation of Rab5 and Rab7 from the membrane, resulting in an elevation of membrane-bound Rabs and a reduction of cytoplasmic Rabs (Fig. 5d). Concurrently, OI-induced redistribution of Rab5 and Rab7 between cytoplasm and membrane was abolished in the absence of GDI2 (Fig. 5d). Therefore, these data indicate that OI inhibits GDI2-mediated extraction of Rabs from the membrane, resulting in enhancement of Rabs’ distribution on the membrane and subsequent viral infection.
Considering that itaconation could hinder protein-protein interactions,6,11 we supposed that itaconation on GDI2 might hinder the interaction between GDI2 and Rabs, resulting in a decreased extraction of Rabs from membrane. Indeed, OI attenuated the interaction between Rabs and GDI2, but have no effect on its interaction with GDI1 (Fig. 5e, f). Itaconate-alkyne (ITalk),34 a probe for itaconation, was utilized to enrich proteins modified by itaconate. The enrichment of GDI2 was observed in the ITalk-labeled proteins (Fig. 5g) from 293 T cells transfected with GDI2-Myc, which could be attenuated by the addition of OI (Fig. 5h), suggesting that both ITalk and OI could alkylate GDI2 and target the same sites. Furthermore, the enrichment of endogenous GDI2 was also observed in the ITalk-labeled proteins from MLE-12 cells (Fig. 5i). The sites of itaconation on GDI2 were subsequently identified, revealing the presence of five alkylated sites (C202, C203, C317, C335, C414), among which three sites (C203, C335, C414) exhibited stable detectability across various experiments (Fig. 5j). These data indicate that GDI2 is a direct target for itaconation.
To identified the critical itaconation sites on GDI2 that contributed to the enhancement of viral infection induced by OI, GDI2-knockout cells were transfected with wildtype GDI2, mutant GDI2 or an empty vector. In the absence of GDI2, OI-induced viral overgrowth was completely abolished (Fig. 5k). Transfection of both wildtype and mutated GDI2 suppressed viral infection; however, only wildtype GDI2 reinstated the enhancing effect of OI on viral infection (Fig. 5k). The C203/C335/C414 mutation of GDI2 was found to be sufficient for blocking the effect of OI (Fig. 5k), demonstrating the indispensability of these three sites within GDI2. The collective data indicate that the addition of itaconate lead to the itaconation of C203/335/414 residue on GDI2, thereby disrupting the interaction between GDI2 and Rabs, increasing Rabs’ membrane distribution, and finally promoting viral overgrowth.
Finally, mice were intranasally administered with adeno-associated virus (AAV) containing GDI2 shRNA to establish a knockdown model of GDI2 (Fig. 5l). Knocking down GDI2 promoted viral infection in lung tissues (Fig. 5m, n), while also abolished the promotion of VSV and IAV infection caused by OI treatment (Fig. 5m, n). These findings indicate that GDI2 inhibits VSV and IAV infection, whereas OI facilitates viral infection through its modification of GDI2 in vivo.
Viral infection induces robust IRG1-itaconate axis activation in neutrophilsNeutrophils-derived itaconate attracts increasing attention in recent years, playing a pivotal role in cancer,13,35 trauma36 and bacterial infection.37 We next identified the cell types responsible for itaconate production during viral infection by single-cell RNA sequencing (scRNA-seq). Observably, neutrophils possessed the highest Irg1 expression (Fig. 6a–c) upon viral infection. Macrophages and monocytes also exhibited moderate Irg1 expression, albeit to a significantly lesser extent than neutrophils (Fig. 6c). Among macrophages, interstitial macrophages exhibited certain level of IRG1 expression, whereas alveolar macrophages demonstrated minimal expression (Fig. 6c). These data were further confirmed by sorting these cell subpopulations post infection (Fig. 6d and Supplementary Fig. 10a). Furthermore, immunofluorescence analysis showed a widespread co-staining of IRG1 with neutrophils (Fig. 6e) and a slight co-staining with macrophages in the lung (Supplementary Fig. 10b).
Fig. 6
留言 (0)