
記住我
Acute pancreatitis (AP) is a common condition in the digestive system, characterized by the abnormal activation of pancreatic enzymes that can lead to inflammation in the pancreas and surrounding organs (1). The annual incidence rate of AP varies from 4.9 to 73.4 per 100,000 individuals, depending on factors such as regional differences, variations in diagnostic criteria, and population demographics, with a rising trend (2, 3). For instance, higher incidence rates are often reported in Western countries due to lifestyle factors such as alcohol consumption and gallstone prevalence, whereas lower rates are observed in regions with different dietary and health risk profiles (4). Additionally, improvements in diagnostic techniques and greater awareness of the disease in recent decades have contributed to the rising trend in AP incidence globally (3, 5). While most patients experience mild symptoms, 20% to 30% may progress to severe acute pancreatitis (SAP), which often involves organ dysfunction. In SAP cases, the mortality rate can be as high as 20% to 40%, posing a significant threat to individuals’ lives and well-being (6). Pancreatic damage typically originates in acinar cells, the primary cell type of the exocrine pancreas, which leads to inflammation as a secondary process. AP is commonly associated with elevated levels of digestive enzymes in the blood, such as amylase and lipase, though these are not strict requirements for diagnosis. Premature activation of digestive enzymes (such as the conversion of trypsinogen to trypsin), formation of large vacuoles within acinar cells, and activation of inflammatory mediators are also key features of the disease. While hyperamylasemia is frequently observed, lipase elevation is considered more specific for diagnosing AP (7, 8). This includes the key transcription factor nuclear factor kappa B (NF-κB), which triggers the infiltration of inflammatory cells in the pancreas and systemic inflammatory response, leading to acinar cell death through apoptosis and necrosis (9). A significant body of research has explored the signaling pathways involved in these pathological processes, elucidating the structure of many molecules that mediate inflammatory responses (e.g., NF-κB, cytokines/chemokines, adhesion molecules, and novel protein kinase C subtypes) and cell death responses (e.g., cysteine enzymes) (10–12). However, other areas of AP research, such as the impact of damage to intracellular organelles like mitochondria, remain incompletely understood, warranting further investigation.
Recent studies have highlighted the importance of intracellular organelles, particularly mitochondria, in the progression of AP. Mitochondrial dysfunction, characterized by impaired ATP synthesis and increased production of reactive oxygen species (ROS), has been implicated in exacerbating pancreatic inflammation and cell death.
Activation of pattern recognition receptors (PRRs) in both immune and non-immune cells often trigger inflammation. These receptors can be activated not only by viruses and bacteria, known as microbe-associated molecular patterns or pathogen-associated molecular patterns, but also by endogenous molecules known as damage-associated molecular patterns (DAMPs) (13). Under normal conditions, DAMPs such as nucleic acids, ATP, and calreticulin are unable to stimulate PRRs due to limited access to subcellular regions where PRRs are located. During inflammation, however, changes in membrane permeability allow these molecules to activate PRRs and drive the inflammatory process (14). Various mitochondrial elements and metabolites can act as DAMPs, potentially triggering inflammatory responses upon their release into the cytosol or the external environment. Research has demonstrated that during AP, changes in membrane permeability of acinar cells and organelles result in mitochondrial DNA (mtDNA) being released and the NLRP3 inflammasome becoming activated, thereby driving inflammation (15).
Research on the pathophysiological mechanisms of AP has advanced, but the precise mechanism of cell damage during inflammation remains unclear, hindering effective clinical treatments. Mitochondria, as the primary energy producers in cells, play a crucial role in cellular homeostasis and survival. Dysfunctional mitochondria contribute to the pathogenesis of AP by disrupting ATP synthesis, increasing ROS production, and altering calcium homeostasis. Studies indicate that intracellular calcium overload in AP leads to mitochondrial dysfunction, disrupting ATP synthesis and causing acinar cell damage and necrosis, exacerbating inflammation (16–18). The severity of AP correlates with necrosis extent, with mitochondria influencing autophagy and apoptosis regulation. This review explores mitochondria’s physiological role in pancreatic function, emphasizing their dysfunction’s role in AP through mechanisms such as calcium overload, ATP depletion, oxidative stress, and membrane permeability changes. Additionally, it also discusses mitochondria-related signaling pathways in AP and proposes potential therapeutic strategies targeting mitochondrial dysfunction for improved AP diagnosis and treatment.
2 Mitochondria: structure, origin, and the role in cellular function and pathophysiologyMitochondria are crucial organelles in eukaryotic cells, often recognized as the cell’s powerhouse due to their critical role in energy production. Structurally, they can be categorized into four distinct regions: the outer mitochondrial membrane (OMM), the mitochondrial intermembrane space, the inner mitochondrial membrane (IMM), and the mitochondrial matrix, each contributing to the organelle’s unique and complex functionality (19). These structures are integral to the mitochondria’s ability to support cellular functions, particularly in energy production through oxidative phosphorylation. Mitochondria are the primary sites for oxidative phosphorylation and ATP production within cells, providing energy for cellular activities (20). The tricarboxylic acid (TCA) cycle breaks down the carbon substrate of acetyl-CoA, derived from pyruvate, amino acids, and fatty acids, generating carbon dioxide and reducing NAD+ to NADH and FAD2+ to FADH2 (21) (see Figure 1). These molecules then serve as substrates for the respiratory chain, driving ATP production through oxidative phosphorylation. The activity of the rate-limiting enzyme in the TCA cycle is dependent on Ca2+, which aids mitochondria in adjusting to heightened cellular ATP demand.
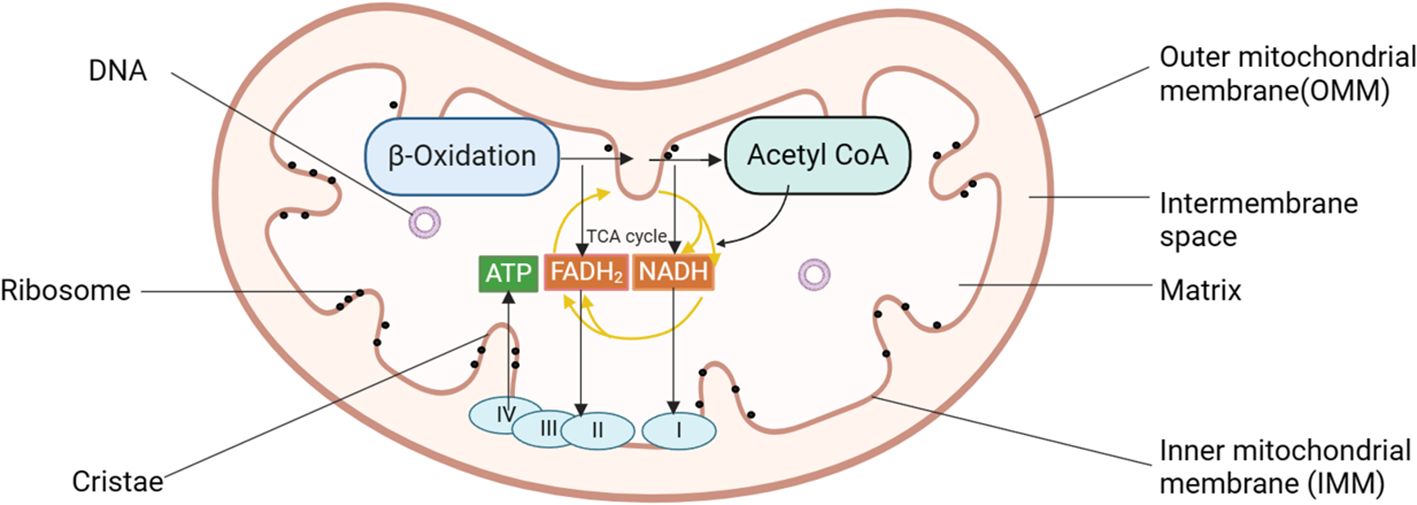
Figure 1. Diagram of mitochondrial structure pattern of pancreatic acinar cells. Mitochondria can be categorized into four distinct regions: the OMM, the mitochondrial intermembrane space, the IMM, and the mitochondrial matrix. Mitochondria serve as the primary sites for oxidative phosphorylation and ATP production within cells, fueling cellular activities. I, complex I, NADH: ubiquinone oxidoreductase; II, complex II, succinate: ubiquinone oxidoreductase; III, complex III, ubiquinol: cytochrome c oxidoreductase; IV, cytochrome c oxidase; ATP, adenosine triphosphate; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane.
Under normal conditions, Ca2+ accumulation in mitochondria remains stable. Ca2+ is released from the ER, triggering zymogen exocrine secretion and promoting ATP production in the mitochondria. However, the rise in cytoplasmic calcium ion concentration is brief as ATP-dependent calcium ion channels swiftly clear the cytoplasmic calcium ions. The sarcoplasmic/endoplasmic reticulum calcium ATPases (SERCAs) move calcium ions into the ER, while plasma membrane Ca2+ ATPases (PMCAs) transport calcium ions out of the cell. Prolonged Ca2+ influx can result in intracellular calcium overload, leading to mitochondrial membrane damage, alterations in mitochondrial membrane potential, and decreased ATP production (22, 23). This disruption is particularly detrimental in the context of AP, where calcium overload accelerates mitochondrial injury and contributes to acinar cell death.
Mitochondria are also essential in producing reactive oxygen species (ROS), which are by-products of ATP generation and have various intracellular effects. Mitochondrial ROS (mitoROS), including reactive ions and molecules, such as superoxide anion (O2-), hydrogen peroxide (H2O2), and hydroxyl radical (-OH), are produced by the electron transport chain in mitochondria. The generation of mitoROS primarily occurs in the mitochondrial respiratory chain during the electron transfer process. While these ROS participate in cell signaling and regulation under normal circumstances, excessive production or inadequate clearance can lead to cellular damage. Low ROS levels can promote cell proliferation. Kirova et al. discovered that mitoROS directly regulate cyclin-dependent kinase 2 (CDK2), targeting a specific conserved cysteine and disrupting the regulatory CDK-related phosphatase (KAP), thus influencing the cell cycle (24). However, elevated mitoROS levels have been linked to pathological conditions, such as pancreatitis, where excessive ROS production induces oxidative stress and triggers apoptosis pathways (25).
Moreover, mitochondria regulate intracellular calcium levels, and their strategic localization within the cell helps compartmentalize calcium signals. Disturbances in calcium homeostasis lead to mitochondrial calcium overload, which disrupts the mitochondrial membrane potential, decreases ATP production, and ultimately triggers acinar cell death through apoptosis and necrosis (26). Thus, the delicate balance between mitochondrial calcium regulation and ROS production is essential for cellular survival, and its disruption is a key driver of pancreatic cell injury in AP.
In pancreatic acinar cells, mitochondria are identified into three distinct groups: perigranular mitochondria at the granular and basolateral boundary, peripheral mitochondria in the basolateral zone near the plasma membrane, and perinuclear mitochondria surrounding the cell nucleus (27–29). These groups of mitochondria play specific roles in regulating intracellular Ca2+ homeostasis. Perigranular mitochondria prevent the diffusion of Ca2+ signals into the basal region, confining physiological Ca2+ signals to the apical region. Peripheral mitochondria supply ATP for Ca2+ pump-mediated uptake into the endoplasmic reticulum (ER) and are implicated in store-operated calcium influx. Perinuclear mitochondria act as a protective shield for the nucleus, protecting it from Ca2+ signal intrusion (30, 31). This spatial organization is essential for maintaining physiological Ca2+ dynamics in pancreatic cells, and its disruption is closely tied to mitochondrial dysfunction in AP.
One significant connection is seen in mitochondrial diseases such as Kearns-Sayre syndrome, where mitochondrial dysfunction directly links to recurrent episodes of pancreatitis, highlighting the impact of disrupted mitochondrial metabolism on pancreatic tissue (32). In AP, mitochondrial injury, particularly through calcium signaling dysregulation, leads to necrosis of pancreatic acinar cells. Calcium overload in mitochondria specifically contributes to necrotic injury, underscoring the importance of maintaining mitochondrial health to prevent acinar cell death (33). Central to this pathology is mitochondrial dysfunction, driving energy deficits and cellular necrosis. This is exacerbated in injured pancreatic acinar cells by impaired macroautophagy, hindering the critical process of clearing damaged mitochondria (34). Studies have emphasized that mitochondrial damage occurs early in the progression of AP, not only affecting the pancreas but also impacting organs like the lungs and jejunum, suggesting that mitochondrial dysfunction contributes to the systemic manifestations of the disease (35). In addition to mitochondrial dysfunction, disorders in calcium metabolism are crucial in promoting cell injury and necrosis in AP. The interplay between mitochondrial calcium overload and energy failure sets the stage for acinar cell death and tissue damage. Given the central role of calcium-mediated mitochondrial dysfunction in AP pathogenesis, therapeutic strategies targeting mitochondrial protection and calcium regulation have been proposed to mitigate disease severity (36). Emerging therapeutic approaches focusing on mitochondrial health offer new hope for AP treatment. Recent research suggests that the delivery of hypoxia-treated functional mitochondria to damaged pancreatic acinar cells by mesenchymal stem cells can alleviate metabolic dysfunction and reduce tissue injury (37). Additionally, mitochondria-targeted therapies, such as Kaempferol nanoparticles, have demonstrated potential in improving mitochondrial homeostasis and reducing inflammation in models of SAP (38).
The distribution of mitochondria within pancreatic ductal epithelial (PDE) cells is still not well understood. Electron microscopy studies in guinea pig PDE cells have shown that mitochondria are most densely packed in the cell’s apical portion (39). The functional importance of this localization remains currently unknown, but it could potentially support the energy requirements for ion secretion across the apical membrane of PDE cells.
3 Mitochondrial dysfunction in acute pancreatitis pathogenesis3.1 Mitochondrial calcium overloadCalcium overload is a significant factor causing acute pancreatitis (AP) in pancreatic acinar cells. Calcium ions (Ca2+) serve as crucial second messengers in cells and act as essential cofactors for multiple digestive enzymes within acinar cells. Proper regulation of Ca2+ levels is vital for sustaining cell functions such as metabolism, proliferation, differentiation, apoptosis, and other cellular processes. The regulation of mitochondrial calcium homeostasis is primarily controlled by the mitochondrial calcium uniporter (MCU), which mediates the uptake of Ca2+ into the mitochondria. During pathological conditions such as AP, MCU becomes hyperactive, leading to excessive Ca2+ accumulation in the mitochondrial matrix, which overwhelms the mitochondria’s buffering capacity. This calcium overload promotes mitochondrial membrane depolarization, impairs ATP synthesis, and triggers cell death through necrotic pathways. A study by M. Chvanov et al. found that knocking out MCU influences mitochondrial Ca2+ dynamics but does not reduce the severity of experimentally induced AP. This suggests that while MCU facilitates mitochondrial Ca2+ uptake, its role in AP pathogenesis may involve compensatory mechanisms or other factors that mitigate its impact (40). Conversely, the mitochondrial Na+/Ca2+ exchanger (NCLX) functions to extrude excess Ca2+ from the mitochondria to prevent calcium overload. Impaired NCLX function has been shown to exacerbate mitochondrial calcium accumulation, further contributing to cellular injury and necrosis in AP (31). This balance between MCU-mediated calcium influx and NCLX-mediated efflux is critical in maintaining mitochondrial integrity and preventing calcium-induced mitochondrial dysfunction (41). Targeting MCU and NCLX to regulate mitochondrial calcium homeostasis presents a promising therapeutic avenue for mitigating mitochondrial dysfunction in AP.
Under physiological conditions, Ca2+ is predominantly stored in the ER, with cytosolic Ca2+ levels approximately 10,000 times lower than those in the extracellular fluid (42). Intracellular calcium levels are generally stable under normal conditions, but studies have shown an increase in intracellular Ca2+ during AP, which correlates with disease severity (43). In normal physiology, Ca2+ acts as a second messenger when released from the ER, triggering the exocrine secretion of zymogen granules (ZG) and promoting ATP production in mitochondria (23, 42). However, the rise in cytoplasmic calcium concentration is transient, as elevated Ca2+ is swiftly removed by two ATP-dependent calcium pumps: the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) pumps Ca2+ back to the ER, while the plasma membrane Ca2+ ATPase (PMCA) expels Ca2+ out of the cell, ensuring intracellular calcium balance (44).
Under pathological conditions, factors such as alcohol, bile acids, and cholecystokinin stimulate the release of Ca2+ from the ER, resulting in intracellular calcium overload. This overload is primarily mediated by the inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) located on the ER membrane (see Figure 2) (45), evidenced that bombesin can enhance the gene expression of IP3Rs and RyRs, causing calcium overload, while docosahexaenoic acid can inhibit this process, thereby restoring normal calcium signaling within acinar cells (46). Sustained release of Ca2+ from the ER depletes calcium stores, triggering stromal interaction molecule 1 (STIM1) to detect the decrease in ER luminal Ca2+ levels and activate store-operated calcium entry (SOCE) channels to replenish calcium stores (47). Calcium release-activated calcium channel protein 1 (ORAI1) plays a crucial role in the SOCE channel. STIM1 is responsible for recruiting and activating ORAI1 to initiate the opening of the SOCE channel (47). The activation of this channel exacerbates calcium overload (48). The ORAI1 inhibitor (CM4620) can prevent acinar cell necrosis and reduce local and systemic inflammatory responses in both human pancreatic acinar cells and animal models of AP (49).
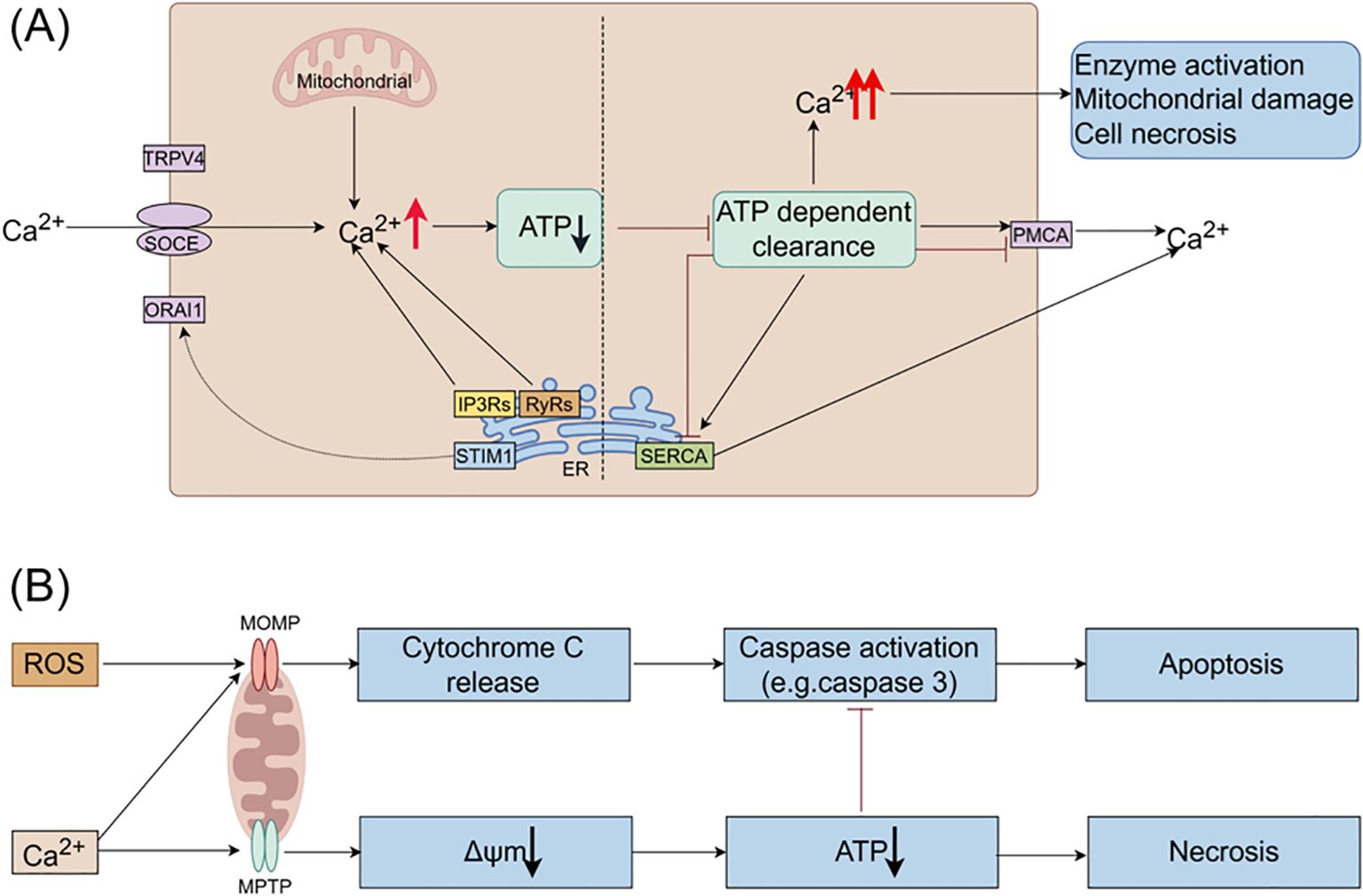
Figure 2. The roles of Ca2+ and ROS in mitochondrial pathways of apoptosis and necrosis in pancreatitis. (A) In pancreatic acinar cells, intracellular Ca2+ overload resulting from pathological Ca2+ signaling or inadequate clearance by ATP-dependent mechanisms is a major factor contributing to the development of acute pancreatitis. The regulation of Ca2+ signals in pancreatic acinar cells involves various Ca2+ channels, such as Ca2+ release channels like IP3Rs and RyRs on intracellular stores, SOCE on the cell membrane, and SERCA for refilling intracellular stores. (B) Ca2+ stimulate the opening of mitochondrial MPTP, resulting in a decrease in mitochondrial membrane potential, ATP depletion, and cell necrosis. ROS promote the release of cytochrome c through MOMP, leading to caspase activation and apoptosis. Additionally, Ca2+ itself can trigger the release of cytochrome c and induce cell apoptosis. Moreover, decreased ATP production inhibits caspase activation. Therefore, mitochondrial depolarization not only mediates necrosis but also restricts apoptosis in pancreatitis, elucidating the inverse relationship between acinar cell necrosis and apoptosis observed in experimental pancreatitis models. ATP, adenosine triphosphate; MOMP, mitochondrial outer membrane permeabilization; MPTP, mitochondrial permeability transition pore; IP3Rs, inositol 1,4,5-trisphosphate receptors; ORAI1, Orai calcium release-activated calcium modulator 1; PMCA, plasma membrane Ca2+ ATPase; ROS, reactive oxygen species; RyRs, ryanodine receptors; SERCA, sarcoplasmic/endoplasmic reticulum calcium ATPase; SOCE, store-operated Ca2+ entry channels; STIM1, stromal interaction molecule 1; TRPV4, transient receptor potential vanilloid 4.
Excessive intracellular Ca2+ levels trigger the opening of the mitochondrial permeability transition pore (MPTP), resulting in the loss of mitochondrial membrane potential and hindering ATP production (50). Consequently, perigranular mitochondria are unable to regulate the rise in apical Ca2+ concentration, leading to the propagation of local Ca2+ signaling across acinar cells (51). Reduced ATP production impairs the ability of SERCA and PMCA to eliminate cytoplasmic Ca2+, thereby perpetuating the escalation of intracellular Ca2+ levels. Prolonged Ca2+ overload induces alterations in mitochondrial membrane permeability, mitochondrial impairment, and ultimately, acinar cell necrosis through a destructive cycle.
In addition, ERCP, cholelithiasis, and other factors can also contribute to AP by triggering the Piezo1 channel in response to high pressure in the pancreatic duct, leading to an influx of Ca2+ (52). Piezo1, present in various tissues, is selective for Ca2+ and can be activated by mechanical pressure, playing a role in pressure-induced AP (53). Research indicates that the activation of Piezo1 channels results in a transient increase in Ca2+ levels in acinar cells, followed by the activation of the transient receptor potential vanilloid 4 (TRPV4), leading to sustained Ca2+ influx. Knocking out the TRPV4 gene in mice has been shown to prevent both Piezo1 agonist-induced and pressure-induced AP (54).
Given the pivotal role of calcium overload in the pathogenesis of AP, targeting calcium channels represents a promising strategy for early intervention. Understanding the intricate calcium signaling in pancreatic acinar cells can lead to the development of therapeutic approaches aimed at modulating calcium homeostasis to prevent or mitigate AP.
3.2 Mitochondrial ATP depletionMitochondria are essential for supplying energy for cellular functions through the synthesis of ATP. Dysfunction in mitochondria can result in ATP depletion, leading to various physiological dysfunctions that rely on ATP, such as ZG secretion, Ca2+ clearance, and autophagy. Continuous calcium influx can cause intracellular calcium overload, damaging the mitochondrial membrane, opening the MPTP, altering the mitochondrial membrane potential, and ultimately reducing ATP production (50). This decrease in ATP levels inhibits ATP-dependent transport mechanisms like SERCA and PMCA, preventing the removal of intracellular Ca2+ and resulting in sustained intracellular calcium overload (see Figure 2). This overload can activate digestive ZG, resulting in the pancreatic autodigestion.
Research has demonstrated that elevated levels of non-conjugated chenodeoxycholate (CDC) can lead to mitochondrial damage and ATP depletion, which in turn can directly hinder the secretion of pancreatic duct fluid and bicarbonate (HCO3−) (39). Studies have shown that supplementing ATP in vitro can prevent damage and dysfunction in acinar cells during AP (55, 56). Furthermore, the MPTP inhibitor cyclosporin A (CsA) and its derivative TRO40303 have been found to decrease acinar cell necrosis and mitochondrial damage, ultimately improving alcoholic AP by inhibiting the opening of MPTP (57). Moreover, early-stage supplementation of high-calorie nutritional support to boost ATP levels may provide a promising approach to mitigating disease progression (58).
3.3 Mitochondrial permeability transition pore dysfunctionThe mitochondrial permeability transition (mPT) is a process that increases the permeability of the IMM in a Ca2+-dependent and cyclophilin D (CypD)-promoted manner, allowing molecules of approximately 1.5 kDa to pass through (59, 60). This phenomenon is regulated by the MPTP, a non-selective channel on the IMM that is sensitive to CsA. Activation of MPTP can occur in response to Ca2+ overload, ROS, and other stress signals, leading to cell death through necrosis or apoptosis.
The pore-forming structure of mPT is currently not well understood. Research has indicated that the channel of mPT may involve the voltage-dependent anion channel (VDAC) (61), pro-apoptotic Bcl-2 family members (Bax and Bak) (62–64), phosphate carrier (PiC) (65), and translocator protein (TSPO) (66). While the exact molecular mechanism of MPTP formation remains uncertain, recent studies suggest that mitochondrial F1F0-ATP synthase, ANT, and the matrix CypD play a role in promoting its transition to a pore-forming conformation (67–69).
MPTP exhibits two distinct opening states: transient and sustained. The transient opening of MPTP regulates various processes including mitochondrial Ca2+ efflux (70, 71), ROS signaling (72), cell metabolism (73), and the differentiation of neurons, cardiac muscle, and stem cells (74–76). This physiological process involves the rapid exchange of solutes between the cytoplasm and the mitochondrial matrix, which is crucial for cellular signaling. However, the balance of MPTP opening is delicate and highly regulated. While transient opening of the MPTP supports essential cellular functions, the sustained opening of the MPTP results in mitochondrial swelling and mitochondrial OMM rupture, leading to subsequent apoptosis and necrotic cell death (77). This process is associated with a series of pathologies. Firstly, the increased permeability of the mitochondrial membrane caused by the continuous opening of MPTP results in the disruption of membrane potential and interruption of electron transport, resulting in diminished ATP production. Insufficient ATP affects the energy supply of cells and exacerbates pancreatic cell dysfunction. Secondly, MPTP opening may increase ROS production. When MPTP opens, oxidative substances and free radicals in mitochondria can escape into the cytoplasm, triggering oxidative stress reactions and causing increased cell damage and inflammatory responses. Additionally, increased mitochondrial membrane permeability can activate apoptotic pathways. The release of apoptosis-related proteins such as Cytc from mitochondria into the cytoplasm upon MPTP opening activates the caspase family, ultimately triggering apoptosis (78, 79). Studies have indicated that in pancreatitis, MPTP opening and increased mitochondrial membrane permeability are crucial mechanisms leading to ATP depletion and cellular dysfunction (50). Finally, MPTP opening is linked to the mtDNA release during innate immunity (80). Research has demonstrated that oxidative stress can induce the release of mtDNA through MPTP in rat liver cells (81).
Increased mitochondrial membrane permeability is a significant contributor to the pathogenesis of pancreatitis. This phenomenon results in decreased energy production, elevated ROS levels, and initiation of apoptosis, ultimately exacerbating pancreatic cell injury and triggering inflammatory processes. Consequently, modulating alterations in mitochondrial membrane permeability could offer a promising therapeutic approach for managing pancreatitis.
3.4 Oxidative stressOxygen free radicals are significant contributors to the pathogenesis of various inflammatory diseases and are crucial in the oxidative stress (OS) mechanism of AP (82). OS occurs when the production of ROS outweighs the capacity of antioxidant defenses, leading to cell damage either directly or by modulating signaling pathways (83). Research has demonstrated that oxygen free radicals are essential in driving the development of AP in an in vivo model of experimental AP (56).
The generation of mitoROS primarily occurs during the electron transfer process of the mitochondrial respiratory chain (84). MitoROS production involves several key steps: (1) Complexes I, II, and III of the respiratory chain: Complex I (NADH dehydrogenase) and complex II (succinate dehydrogenase) on the IMM convert H+ to coenzyme Q, releasing electrons in the process. These electrons are then transferred to Cytc through complex III. (2) Cytc redox: Cytc passes on the electrons to complex IV of the respiratory chain. (3) Complex IV: Complex IV accepts the electrons and combines them with oxygen to produce water. However, some electrons may escape the respiratory chain during this process and react with molecular oxygen to generate O2- (85, 86). While O2- itself is relatively inactive, it can lead to the formation of more harmful ROS, such as hydrogen peroxide (H2O2) and hydroxyl radicals (-OH), through subsequent reactions.
In AP, the pancreas releases inflammatory mediators that place mitochondria under sustained high load, leading to damage to the mitochondrial respiratory chain. This dysfunction, along with the excessive accumulation of ROS, is key in the progression of the disease. The impaired mitochondria hinder the activities of MAPK and AKT, resulting in insufficient ATP production to sustain cellular functions and exacerbating ROS accumulation.
ROS is essential in regulating the inflammatory response in AP. This inflammatory response is a key pathological process in pancreatitis, and the overproduction of mitoROS can activate various inflammatory signaling pathways, including NF-κB and NLRP3 inflammasome. Activation of these pathways leads to increased inflammatory cytokines release, including TNFα and IL-1β, thereby perpetuating and exacerbating the inflammatory response (87). Furthermore, mitoROS are implicated in pancreatic cell damage and necrosis in AP. Mitochondrial dysfunction induced by pancreatitis leads to heightened production of mitoROS, causing elevated intracellular oxidative stress. The ROS generated during OS can directly harm cell membranes, nucleic acids, and proteins, ultimately triggering cell apoptosis and necrosis (88–90).
Studies have shown that in AP, mitoROS production initiates cell apoptosis and disrupts insulin secretion function by activating the apoptosis signal-regulated kinase 1 (ASK1) pathway (91). Additionally, mitoROS contribute to the development of pancreatic fibrosis in pancreatitis. The chronic progression of pancreatitis can result in pancreatic fibrosis, with excessive mitoROS production closely linked to fibrosis formation. MitoROS can enhance the activation of pancreatic stellate cells, prompting the synthesis of extracellular matrix and collagen deposition, thus altering pancreatic tissue structure and fostering fibrosis. Moreover, ROS may also affect the stability of mtDNA and the transmission of genetic information, influencing gene expression and function in pancreatic cells. MtDNA is vulnerable to direct attack by ROS, leading to oxidative damage and mutations that disrupt mitochondrial function. This further exacerbates the abnormal state of mitochondria and the generation of ROS, creating a vicious cycle. Studies have shown that in an AP model, mitochondrial function is impaired due to inflammatory reactions, resulting in a significant increase in intracellular ROS levels. These ROS can activate the NF-κB signaling pathway, promoting the production of inflammatory cytokines, leading to the persistence and exacerbation of inflammatory reactions (92). Additionally, excessive ROS production triggers mitochondrial membrane depolarization and the activation of apoptotic signals, causing necrosis and apoptosis of pancreatic cells (93).
The roles and impacts of mitoROS in pancreatitis are diverse. It plays a part in regulating inflammatory responses, causing pancreatic cell damage and necrosis, contributing to pancreatic fibrosis, and disrupting normal pancreatic function through effects on mitochondrial function and cellular gene expression. A comprehensive understanding of how mitoROS functions is crucial for uncovering the pathogenesis of pancreatitis and developing effective treatment approaches.
3.5 Impaired autophagyAutophagy is a fundamental biological mechanism where lysosomes are used to break down macromolecules and damaged organelles. It is controlled by autophagy-related genes (Atg) (94). Through autophagy, cells can eliminate, recycle, and degrade various defective cytoplasmic contents, such as damaged organelles, denatured proteins, or lipids, to prevent ER stress and maintain protein synthesis. This process plays a crucial role in sustaining cell homeostasis (95, 96). Research has indicated that impaired autophagy is also a contributor for the pathogenesis of AP (97).
The process of autophagy involves several steps based on the pathways of substrates entering lysosomes, including macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Initially, upon cell stimulation by signals, the UNC-51-like kinase 1 (ULK1) complex is detached from and activated by the mammalian target of rapamycin complex 1 (mTORC1) through a cascade reaction, thereby initiating autophagy (98). Subsequently, the activated ULK1 complex recruits the Beclin1-Vps34 complex, resulting in the creation of double-layered intracellular membrane vesicles. These vesicles, containing various Atg-encoded proteins and ubiquitination receptors (such as p62), elongate with the help of microtubule-associated-protein light-chain-3-II (LC3-II), encapsulate waste products, and give rise to autophagosomes (99, 100). Finally, the autophagosomes merge with lysosomes, a process facilitated by lysosome-associated membrane proteins (LAMPs). Upon fusion, lysosomal cathepsins like CTSB and CTSL degrade and clear the waste material, allowing for recycling (101, 102).
Autophagy impairment in AP acinar cells is characterized by elevated autophagosome production and decreased lysosomal degradation, leading to heightened inflammatory cell infiltration, acinar cell necrosis, and apoptosis (103). Studies have shown that impaired autophagy contributes to acinar cell vacuolization and zymogen activation, triggering AP development (104). Lysosome-associated membrane protein-2 (LAMP-2), a lysosomal membrane protein abundant in pancreatic tissue, plays a crucial role in autophagosome-lysosome fusion (105). In an acute necrotizing pancreatitis rat model, decreased levels of LAMP-2 led to the accumulation of undegraded material in abnormally enlarged vacuoles within acinar cells, indicating impaired autophagy (106). Knockout of LAMP-2 hindered autophagosome-lysosome complex formation, resulting in the buildup of autophagosomes, limited zymogen granule degradation, and abnormal trypsinogen activation, exacerbating AP (107, 108). Additionally, knockout of Atg5 and Atg7 in mouse models of AP impaired autophagy and worsened the disease (109). LC3, the earliest autophagy marker identified, is closely linked to autophagosome abundance, serving as a key indicator of autophagy activity (110). Moreover, p62, a well-studied autophagy substrate, plays a critical role in mitochondrial clearance (111). The impairment of autophagy efficiency in AP can be observed through elevated levels of pancreatic autophagy markers LC3-II and autophagy substrate p62/SQSTM1, as well as an increase in ubiquitinated protein accumulation (112). Research indicates that in AP, interventions targeting the modulation of LC3 and p62/SQSTM1 expression can enhance autophagy and mitigate pathological harm to the pancreas (113, 114).
The process that selectively eliminates damaged mitochondria is known as mitophagy. Mitophagy plays a crucial role in the pathogenesis of pancreatitis by regulating cellular processes that include inflammation and cell death. Impairments in mitophagy may result in insufficient removal of damaged mitochondria, leading to exacerbated pathological responses in pancreatitis, such as a shift in the balance between apoptosis and necrosis, which are pivotal in determining the severity of pancreatitis (115). When mitochondria lose their membrane potential, it can trigger the onset of autophagy, particularly mitophagy, creating a harmful cycle (116).
Mitophagy is primarily regulated by several key pathways, including the PINK1/Parkin pathway, the Bnip3/Nix-mediated pathway, and the FUNDC1-mediated pathway. In the PINK1/Parkin pathway, PINK1 accumulates on the outer mitochondrial membrane when the membrane potential is lost, leading to the recruitment of the E3 ubiquitin ligase Parkin. Parkin ubiquitinates mitochondrial surface proteins, marking them for degradation via the autophagosome (117). This pathway plays a critical role in mitochondrial quality control, ensuring the removal of damaged mitochondria to maintain cellular homeostasis, especially in high-energy-demanding cells, such as neurons and acinar cells (118). Studies also highlight the role of the Bnip3 and Nix proteins in facilitating mitophagy by promoting the interaction between damaged mitochondria and the autophagy machinery (119). Furthermore, Bnip3 can suppress PINK1 cleavage, enhancing the accumulation of full-length PINK1, which is necessary for efficient Parkin recruitment and mitophagy activation (120). The FUNDC1-mediated mitophagy pathway, particularly active under hypoxic conditions, plays a critical role in maintaining mitochondrial homeostasis. FUNDC1 is a mitochondrial outer membrane protein that interacts with LC3 to facilitate the clearance of damaged mitochondria (121). Hypoxia and mitochondrial dysfunction trigger the dephosphorylation of FUNDC1, enhancing its interaction with LC3 and promoting mitophagy, which helps protect cells during stress conditions such as AP (122). Additionally, mitochondrial damage can trigger inflammatory responses, worsening the situation.
Therefore, suppressing the formation of autophagosomes can help mitigate damage caused by autophagy, decrease the activation of digestive enzymes, and provide some relief from AP (123). Pharmacological manipulation of autophagy presents a viable avenue for treating AP. Research indicates that IL-22 can mitigate autophagosome formation via the Beclin1 pathway, thus alleviating the severity of AP (124). The disaccharide trehalose has demonstrated enhanced autophagic efficiency and reduced pancreatic damage in animal models of AP, suggesting its potential as a therapeutic agent (2). Moreover, lycopene has shown promise in ameliorating AP severity in mice by modulating autophagy; however, further investigation is warranted to elucidate its mechanism of action and therapeutic efficacy in human pancreatitis (125). Notably, studies suggest that statins may lower the incidence of AP and enhance prognosis (126, 127). In a rat model of AP, simvastatin restored autophagic flux by promoting autophagosome-lysosome fusion, thereby mitigating mitochondrial damage and inflammatory responses (101). This further supports the notion that promoting complete mitophagic flux, rather than just the initiation of mitophagy, could be critical in mitigating pancreatic inflammation and damage in AP. Thus, understanding the specific mechanisms of mitophagy, particularly the regulation of PINK1/Parkin, Bnip3/Nix and FUNDC1 pathways, may lead to novel therapeutic targets in AP (128). Additionally, the balance mitophagy maintains between apoptosis and necrosis underscores its significance in the pathology of pancreatitis, particularly in relation to inefficient lysosomal function and autophagy impairment (115). Consequently, exploring the function and control of autophagy in AP, as well as developing interventions to restore autophagy in AP acinar cells, are poised to be pivotal areas of future research.
3.6 Mitochondrial regulation of cell death (apoptosis, necrosis and pyroptosis)Besides supplying energy to cells, mitochondria regulate necrosis and apoptosis of acinar cells through changes in mitochondrial membrane permeability (MMP) (19). Excessive Ca2+ accumulation in acinar cells triggers the opening of the MPTP in the IMM, leading to loss of mitochondrial membrane potential, impaired ATP production, and eventual necrosis (see Figure 2). The Bcl-2 family proteins play a key role in mediating the release of Cytc and regulating apoptosis by controlling mitochondrial outer membrane permeability (MOMP) (129). During AP, both necrosis and apoptosis occur simultaneously in acinar cells.
Acinar cell death is a key pathological response in AP. Research has demonstrated that in animal models of AP, the severity is directly proportional to the extent of necrosis and inversely proportional to the level of apoptosis. Furthermore, inducing acinar cell apoptosis has been shown to decrease the severity of necrosis and AP, whereas inhibiting apoptosis with caspase inhibitors like XIAP can worsen necrosis (130). Hyperbaric oxygen therapy has been found to alleviate disease severity by promoting acinar cell apoptosis and reducing necrosis (131). Using isolated mitochondria and acinar cells, it was found that increased expression of the Bcl-2 protein can decrease pancreatic acinar cell necrosis by preventing mitochondrial depolarization and subsequent ATP depletion (132). Additionally, silencing the hypoxia-inducible factor 1α (HIF1α) gene can enhance intracellular energy balance by preserving mitochondrial homeostasis, reducing necrosis, and promoting apoptosis, ultimately mitigating the inflammatory response in AP (133).
Furthermore, mitochondrial dysfunction is recognized as a significant factor in the pyroptosis of pancreatic acinar cells during AP. Mitochondrial damage can result in elevated intracellular Ca2+ levels, which in turn induces OS within the cell. This OS may lead to cell membrane rupture and subsequent pyroptosis (134, 135). Lieberman et al. demonstrated that the N-terminal pore-forming fragment of Gasdermin D (GSDMD-NT) targets mitochondria; during the pyroptosis process, GSDMD-NT rapidly damages both IMM and OMM leading to reduced mitochondrial numbers, mitophagy, ROS, loss of transmembrane potential, attenuated oxidative phosphorylation and release of mitochondrial proteins and DNA from the matrix and intermembrane space (136). Research has indicated a reciprocal relationship between mitochondrial dysfunction and inflammasome activation. ROS released from mitochondria can activate the NLRP3 inflammasome, thereby facilitating the onset of pyroptosis. This process establishes a vicious cycle: mitochondrial damage leads to inflammasome activation, which in turn exacerbates mitochondrial damage (137, 138).
Recent research has identified that certain cytokines, such as IL-37, may exert a protective effect in cases of AP. IL-37 has been shown to inhibit the pyroptosis of damaged acinar cells, a mechanism that appears to be linked to its ability to inhibit the activation of the NLRP3 inflammasome. By specifically removing GSDMD from the pancreas, researchers demonstrated that the protective effect of IL-37 was neutralized, indicating that GSDMD plays a crucial role in the pyroptosis process (139). Additionally, drugs targeting pancreatic acinar cell pyroptosis, such as high-density lipoprotein (HDL) and apoA-I, have been found to inhibit both the activation of the NLRP3 inflammasome and pyroptosis in acinar cells (135). This suggests a novel therapeutic approach for the treatment of AP, potentially alleviating the condition by modulating the inflammatory response. Besides, cold-inducible RNA binding protein (CIRP) has been implicated in inducing mitochondrial dysfunction and pyroptosis in pancreatic acinar cells, indicating that blocking CIRP may represent an effective strategy for treating AP (140).
3.7 Mitochondrial dynamics imbalanceMitochondrial dynamics involves the continuous fusion and fission processes that mitochondria undergo to regulate the shape, number, and distribution. This process is crucial for maintaining mtDNA, ATP production, calcium homeostasis, signal transduction, and apoptosis (141, 142). Imbalances in mitochondrial fission and fusion often result in structural changes and dysfunction within mitochondria. Abnormal mitochondrial fusion can cause fragmentation, while impairments in mitochondrial fission can result in the oversized mitochondria formation. These imbalances in mitochondrial dynamics can disrupt the intracellular environment, cause cellular damage, and even result in cell death. Mitochondrial fusion is primarily regulated by mitofusin-1/2 (MFN1/2) and optic atrophy 1 (OPA1), while mitochondrial fission is mainly controlled by dynamin-related protein 1 (DRP1) (see Figure 3) (143–146).
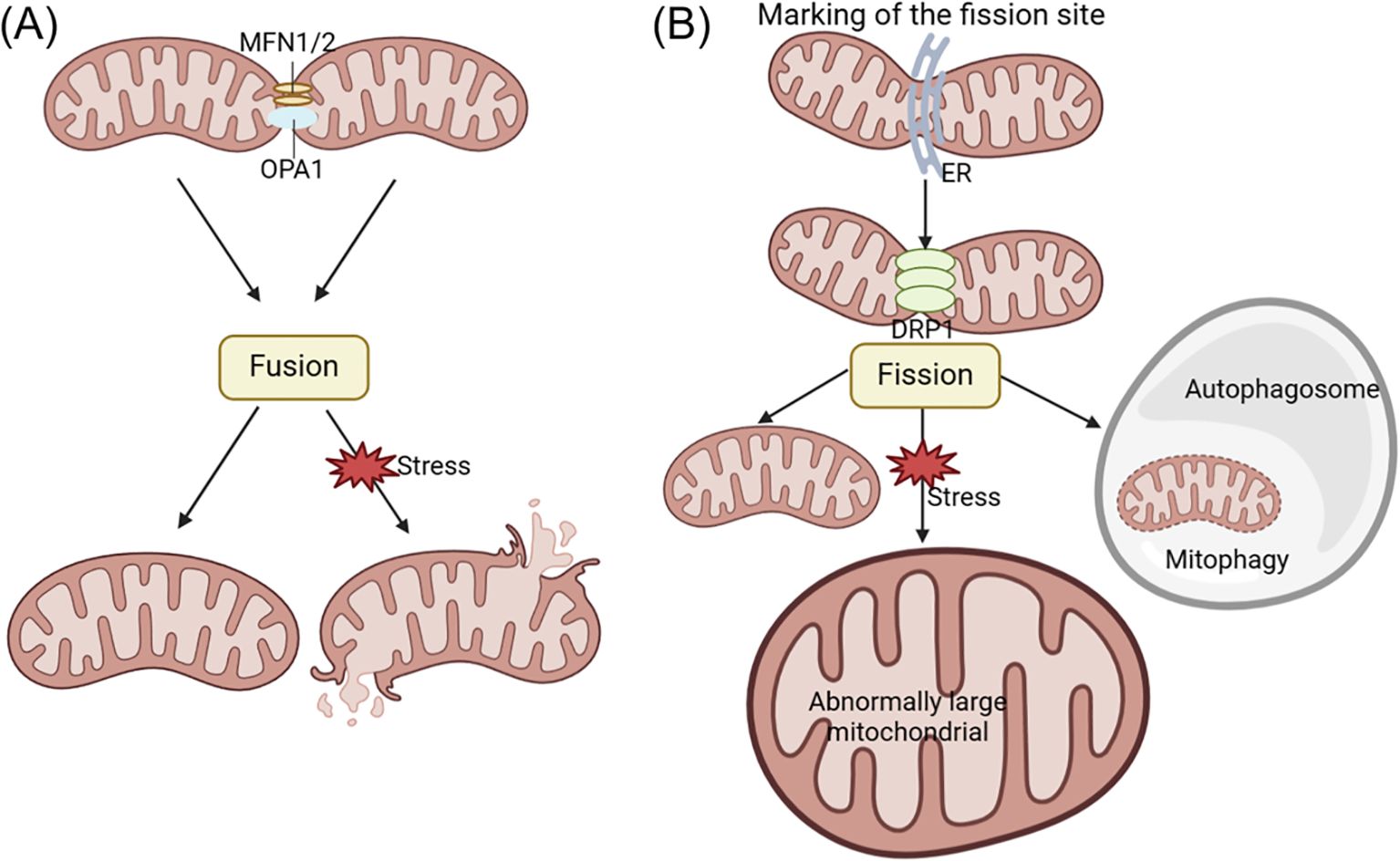
Figure 3. The changes of acinar cell mitochondrial dynamics in pancreatitis. Mitochondrial fusion and fission processes play a crucial role in promoting ATP production and maintaining quality control within the cell. Fusion involves the mixing of mitochondrial contents, while fission generates new healthy mitochondria and facilitates the removal of defective mitochondria through mitophagy. Mitochondrial fusion is primarily regulated by MFN1/2 and OPA1, while mitochondrial fission is mainly controlled by DRP1 regulation. Abnormal mitochondrial fusion can cause fragmentation (A), while disorders in mitochondrial fission can lead to the formation of oversized mitochondria (B). DRP1, dynamin-related protein 1; ER, endoplasmic reticulum; MFN1/2, mitofusin-1/2; OPA1, optic atrophy 1.
Mitochondrial dysfunction during AP development is characterized by disruptions in mitochondrial dynamics, as evidenced by variations in OPA1 and DRP1 expression and distinct ultrastructural features such as mitochondrial fission, elongation, and mitophagy (147). Research has shown that the TAK-242, a novel toll-like receptor 4 (TLR4) antagonist, can protect taurocholate-induced AP acinar cells in mice. TAK-242 was found to prevent changes in protein expression associated with mitochondrial dynamics. Specifically, the levels of OPA1 and MFN1 were elevated compared to the control group of normal mice, while DRP1 expression decreased. These results demonstrate that TAK-242 can enhance cellular function in AP by modulating mitochondrial dynamics and reducing taurocholate-induced cytotoxicity (148).
3.8 Mitochondrial DNA integrity and dysfunctionThe role of mtDNA in pancreatitis spans several critical aspects, ranging from its contribution to the pathogenesis of the disease to its potential as a biomarker for diagnosing disease severity. Numerous studies have highlighted how mitochondrial dysfunction, often influenced by mtDNA alterations, can significantly impact the progression of pancreatitis.
Mutations and dysfunctions in mtDNA have been associated with various pancreatic diseases. For instance, a patient with Kearns-Sayre syndrome, a disorder linked to mtDNA-related mitochondrial dysfunction, experienced recurrent episodes of AP, underscoring the direct impact of mtDNA on pancreatitis (32). Furthermore, mitochondrial dysfunction, including impairments in mitochondrial network dynamics, cristae morphology, and mtDNA nucleoid structure, plays a crucial role in diseases like type 2 diabetes, which affect pancreatic β-cells by disrupting glucose sensing and regulation (149). Although these mechanisms are not directly linked to pancreatitis, they emphasize the broader role of mtDNA in maintaining pancreatic health.
Mitochondrial complex I deficiency has been shown to promote pancreatic α-cell proliferation in models of premature aging, suggesting that mtDNA mutations may have compensatory or pathogenic roles in pancreatic tissue (150). Additionally, VMP1-dependent selective mitophagy and mitochondrial fragmentation, driven by mtDNA, act as protective cellular mechanisms in pancreatitis, highlighting mtDNA’s involvement in cellular responses to the disease (147). Moreover, circulating mtDNA has emerged as a potential biomarker for predicting the severity of AP, indicating that its role extends beyond cellular functions to include disease diagnosis and prognosis (151). This is supported by evidence that mtDNA contributes to mitochondrial dysfunction and induces apoptosis in acinar cells, playing a key role in the pathogenesis of pancreatitis (152).
Beyond its pathogenic influence, mtDNA also plays a role in shaping the course of pancreatitis through mechanisms that regulate cell survival, such as balancing apoptosis and necrosis, which are pivotal in determining the severity of the disease (115). The balance between these processes highlights the critical importance of mitochondrial health in influencing the progression of pancreatic diseases. While numerous studies have identified genetic mutations in nuclear genes, including PRSS1, PRSS2, SPINK1, CFTR, CTRC, CASR, and CLDN2, which are strongly associated with different forms of pancreatitis (153, 154), studies have also explored the potential role of mitochondrial DNA (mtDNA) mutations in the disease’s pathogenesis. For example, the A3243G mutation in mitochondrial DNA, especially within the tRNALeu(UUR) gene, has been identified as a contributing factor to the increased prevalence of diabetes and notably recurrent pancreatitis within a selected familial grouping (155). This suggests that mtDNA mutations may predispose individuals to pancreatitis by impairing pancreatic β-cell function and exocrine regulation. Additionally, a case study reports the first case of chronic pancreatitis associated with mitochondrial encephalopathy, linked to the A8344G mtDNA mutation, highlighting the potential role of mitochondrial dysfunction in recurrent pancreatitis (156). Furthermore, the mtDNA nt7778 G-to-T polymorphism does not exacerbate cerulein-induced AP in mice but may accelerate the progression of autoimmune-like lesions after tissue damage, particularly in older mice, indicating its potential role for the polymorphism in autoimmune disease susceptibility following pancreatic injury (157).
4 Mitochondrial-related signaling pathways in acute pancreatitisMitochondrial dysfunction can cause the release of various components and products, which can trigger inflammatory responses when they accumulate in the cytoplasm or extracellular environment, potentially leading to cell death. Multiple signaling pathways that initiate inflammatory responses as a result of mitochondrial dysfunction have been discovered, particularly cyclic GMP-AMP synthase (cGAS) - Stimulator of Interferon Genes 1 (STING1) and inflammasome signaling pathways (see Figure 4) (158).
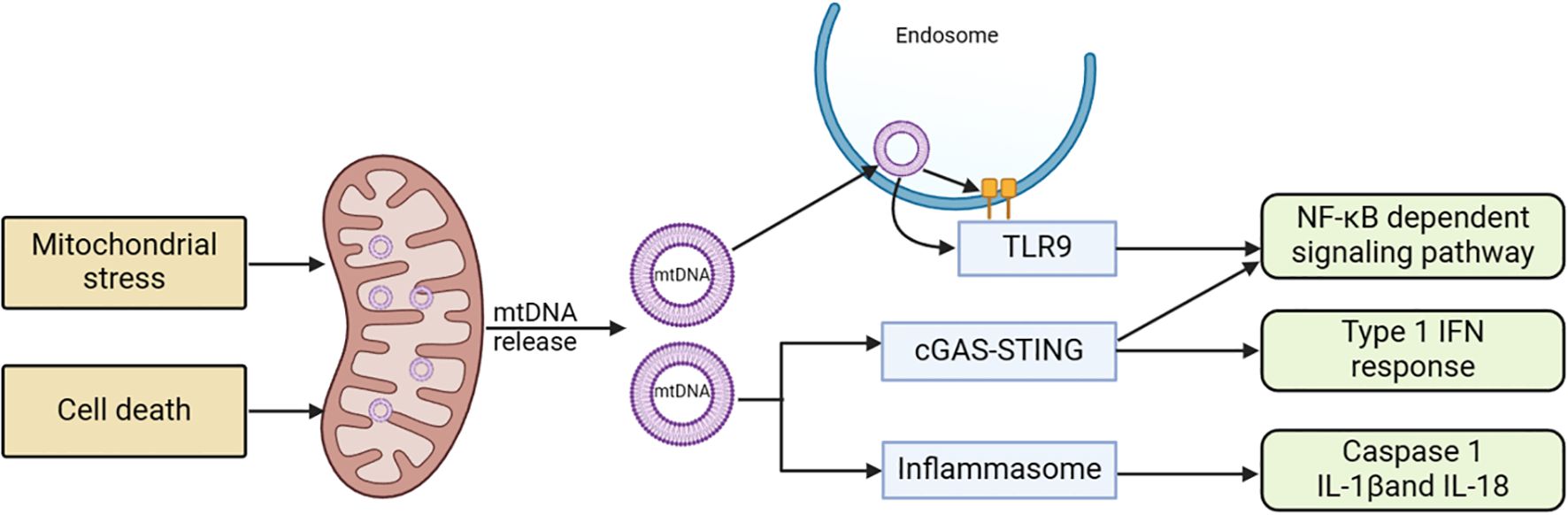
Figure 4. Mitochondria-related signaling pathways and acute pancreatitis. Stress stimulation or cell death leads to the release of mtDNA into the cytoplasm. This mtDNA can initiate various pro-inflammatory signaling pathways either through endosomally localized TLR9, cytoplasmic cGAS-STING, or cytoplasmic inflammasomes. TLR9 binds to mtDNA in endosomes, activating an NF-kB-dependent pro-inflammatory signaling cascade. cGAS detects mtDNA in the cytoplasm and triggers STING, located in the ER, resulting in an interferon response. Inflammasome activity dependent on mtDNA leads to caspase-1 activation or the production of pro-inflammatory IL-1 and IL-18 precursors. cGAS, cyclic GMP-AMP synthase; ER, endoplasmic reticulum; mtDNA, mitochondrial DNA; STING1, stimulator of interferon response cGAMP interactor 1; TLR9, Toll-like receptor 9.
4.1 cGAS–STING1 signaling pathwayThe cGAS-STING is a molecular signaling pathway that involves two proteins: cGAS activated by mtDNA and STING1 (159). cGAS, a cytoplasmic double-stranded DNA (dsDNA) sensor protein, catalyzes the formation of cGAMP (160). Acting as a second messenger that triggers inflammatory responses, cGAMP activates STING1. When mitochondrial outer membrane permeability (MOMP) or other forms of mitochondrial dysfunction led to the entry of mtDNA into the cytoplasm, cGAS signaling is promoted, a process that is hindered by apoptotic caspases (161, 162). In situations where apoptotic caspase activation is limited, mtDNA tends to interact with cGAS and STING1, subsequently resulting in the initiation of type I interferon (IFN) response. Upon exiting the mitochondria, mtDNA can activate cGAS through pores formed by BCL-2 related proteins like BAX and BAK1, or through the permeability transition pore complex (PTPC). This activation leads to the promotion of the STING1 signaling pathway and the expression of inflammatory mediators such as IFN-β1, IL-6, and TNFα (163, 164). While this system usually prevents unnecessary cGAS activation in normal conditions, it retains the ability to trigger inflammatory responses when needed. Numerous studies have demonstrated that mtDNA can strongly induce inflammation via cGAS and STING1, particularly when apoptotic caspase activation is limited (165).
Upon activation, STING1 undergoes a conformational change and translocates to the Golgi apparatus, where it activates downstream kinases, including TBK1 (TANK-binding kinase 1) and IKK (IκB kinase). This results in the phosphorylation and activation of the transcription factors IRF3 and NF-κB (166, 167). Once activated, NF-κB translocates to the nucleus and promotes the transcription of genes encoding inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukins (e.g., IL-1β, IL-6), and chemokines (168). These mediators recruit and activate immune cells in the pancreas, exacerbating the inflammatory response. That is, the pathway induces a positive feedback loop by expressing adhesion molecules and receptors that enhance immune cell infiltration into the pancreas (169). This amplifies the local inflammatory response, potentially leading to pancreatic tissue damage and necrosis.
4.2 Inflammasome signaling pathwayMtDNA and ROS can induce inflammasome activation. In addition to serving as an effective cGAS stimulant, cytosolic mtDNA can trigger inflammasome activation (170). The inflammasome signaling pathway demonstrates that after being released from dysfunctional mitochondria, mtDNA and ROS activate caspase-1, leading to the secretion of IL-1β and IL-18 (171, 172). The electron transport chain (ETC) affects inflammasome activation independently of ROS, maintaining cellular ATP availability through phosphocreatine (173). Here, mtDNA and mitoROS act as the primary DAMPs for inflammasome activation, interacting at multiple nodes in the molecular mechanisms regulating regulated cell death (RCD), thereby significantly influencing RCD (174).
4.3 Other inflammatory pathwaysMitochondrial DNA and other mitochondrial components can also activate inflammatory responses through various PPRs (175). The Toll-like receptor family (TLR) is responsible for detecting a wide range of bacterial signatures to initiate innate immunity. TLR9 is primarily found in monocytes, macrophages, plasmacytoid dendritic cells, and B lymphocytes. While TLR9 is initially located on the ER in its inactive state, it recog
留言 (0)